Practice Essentials
Light-chain deposition disease (LCDD) is the deposition of monoclonal light chains in multiple organs. It is a rare disease characterized by deposition of nonamyloid immunoglobulin light chains, which unlike amyloid do not stain with Congo red and do not exhibit a fibrillar structure when examined ultrastructurally. [1, 2, 3, 4]
LCDD is categorized as a monoclonal immunoglobulin deposition disease in the World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues. [5, 6] LCDD was first described in 1976 in two patients with end-stage renal disease as granular deposits of free light chains in multiple organs, including the kidneys, that did not stain with Congo red. [7]
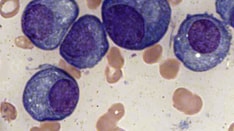
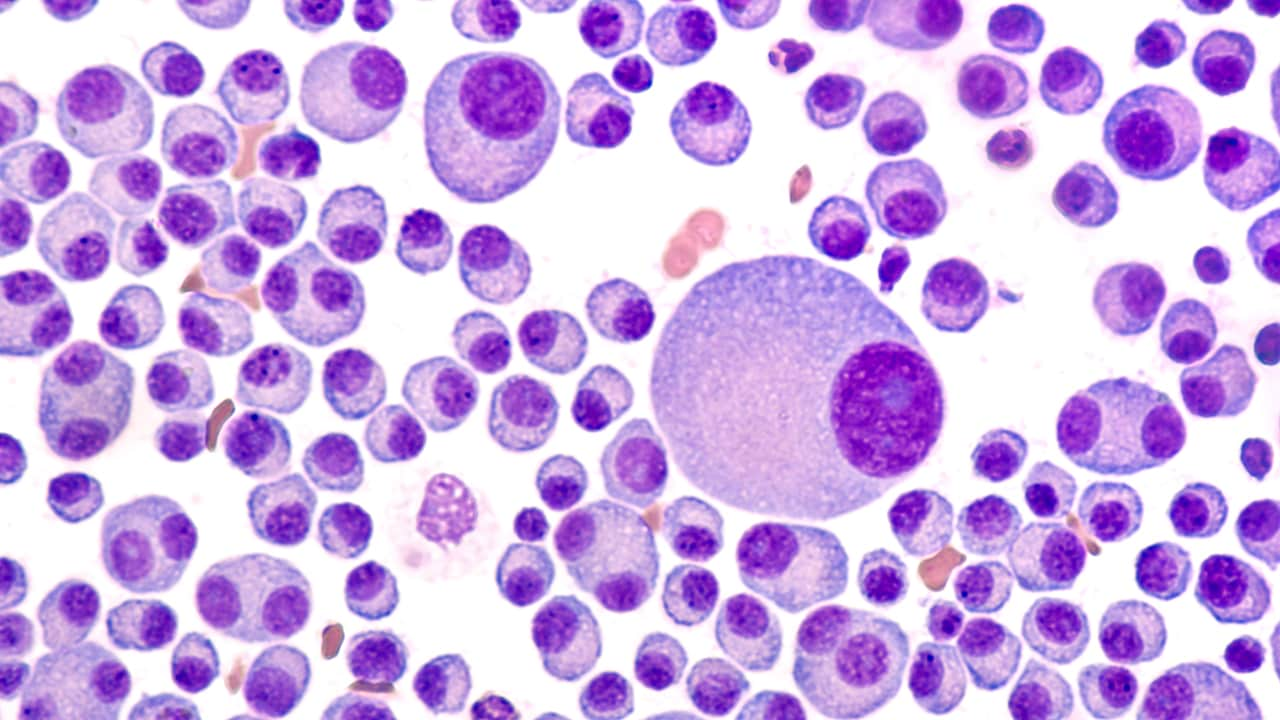
A single clonal plasma cell is responsible for the overproduction of kappa chains and more rarely, lambda light chains. [8] These light chains may be in association with a heavy chain as a complete antibody or be secreted as only a collection of light chains or as a combination of both, [9] although in LCDD only light chains will be observed. A monoclonal population of plasma cells can be detected in the bone marrow, and an altered serum-free light chain ratio is present. [10] In 25% of patients, an abnormal serum free light chain ratio is noted, even without an abnormal finding with serum and/or urine electrophoresis with immunofixation. [11]
The incidence of LCDD is unknown. The median age at diagnosis is 58 years, and it is more common in men than in women. [10] Approximately 50-60% of patients with LCDD have associated multiple myeloma and 17% have monoclonal gammopathy of unknown significance (MGUS) or no evidence of neoplastic plasma cell proliferation. [5]
LCDD can occur in any organ. [12] Kidneys are nearly always involved, and LCDD can present as kidney insufficiency, proteinuria, microscopic hematuria, and nephrotic syndrome. Albuminuria levels do not correlate with the presence of glomerulonephritis and can be present even without glomerulosclerosis. [13] Kidney function declines rapidly as rapidly progressive glomerulonephritis or acute tubulointerstitial nephritis. [5] Extrarenal LCDD most commonly involves the liver but may affect organs with a high degree of vascularity. [14, 15]
Cardiac LCDD has been noted, manifesting in incidents of arrhythmias and even as an atrial mass. [16] Patients with pulmonary LCDD present with dyspnea, hemoptysis and chest pain, though symptomatic cases are rare; more typically pulmonary LCDD is an incidental finding in asymptomatic patients with lymphoproliferative disorders. [17]
The approach for diagnosis with LCDD should be the same as for patients with plasma cell dyscrasias. [18] It should include a thorough history, physical examination, and laboratory and imaging studies (see Presentation and Workup). LCDD should be distinguished from Fanconi syndrome, myeloma cast nephropathy, cryoglobulinemia, and amyloidosis, all of which are also associated with monoclonal proteins and may present similarly.
LCDD is a rare disease, so no established guidelines exist and management remains controversial. [19] Treatment options include the following (see Treatment):
-
Autologous stem cell transplantation (ASCT)
-
Bortezomib
-
Immunomodulatory drugs
-
Daratumumab
-
Kidney transplantation
Pathophysiology
Renal light-chain deposition disease (LCDD) is characterized by the presence of: nodular sclerosing glomerulonephritis on light microscopy; diffuse linear staining in the glomerular and tubular basement membrane by a single light chain on immunofluorescence; and nonfibrillar, powdery, electron-dense deposits in the basements on electron microscopy. [20] Mesangial nodularity within the glomerulus occurs from the increased deposition of extracellular matrix proteins mixed with kappa light chains. [20]
The necessary criterion to make the diagnosis of LCDD requires that all tissues to be stained for kappa and light chains and the tissue must exhibit kappa fixation along the tubular basement membrane. [21, 22, 23] The tubular deposits are present predominantly along the loops of Henle, distal tubules, and proximal tubules.
Amyloid light-chain (AL)–amyloidosis consists predominantly of lambda light chains, whereas kappa light chains are predominantly involved in LCDD. Electron microscopy is helpful in distinguishing between these lesions.
Extrarenal involvement
Symptomatic extrarenal LCDD is rare. [24] The liver is the most frequent site involved. [11, 25] The deposits in the liver are usually confined to the sinusoids and basement membrane of biliary ducts, without associated parenchymal lesions. The degree of liver involvement does not correlate with the amount of light chains deposited. [25] Patients may develop cirrhosis and/or portal hypertension and may die from liver failure. [11]
The cardiac manifestations include restrictive cardiomyopathy, cardiomegaly, congestive heart failure, and arrhythmias. [26, 27, 28] Echocardiography and cardiac catheterization may reveal diastolic dysfunction and decreased myocardial compliance. [29]
Pulmonary LCDD is rare, and it usually damages the lung parenchyma. Involvement of the large airways has been reported. [30] Nodular and diffuse pulmonary interstitial diseases of the lungs have been described. [31, 32, 33]
LCDD can affect peripheral nerves, resulting in polyneuropathy. [34] Isolated LCDD restricted to the brain has been reported as well, wherein the periventricular foci of intracerebral vessels were overloaded with amorphous, eosinophilic material that stained for lambda light chains. [35] In general, the blood-brain barrier protects the central nervous system (CNS) from circulating, misfolded proteins, but cases of intracerebral amyloidoma have been reported. [36]
LCDD can also affect other sites, such as lymph nodes, skin, spleen, pancreas, bone marrow, thyroid gland, adrenal gland, and abdominal blood vessels. [10]
LCDD is associated with multiple myeloma is about 58% of cases. [5] LCDD could be present at diagnosis of a new plasma cell dyscrasia or could represent an extramedullary manifestation of multiple myeloma while relapsing after chemotherapy. [29] LCDD can also complicate other B-cell lymphomas, such as lymphoplasmacytic lymphoma, marginal-zone lymphoma, and chronic lymphocytic lymphoma. [37]
Epidemiology
Frequency
United States
The frequency of light-chain deposition disease (LCDD) is unknown. The disease is found in approximately 5% of patients with multiple myeloma at autopsy.
Mortality/Morbidity
The most common cause of mortality and morbidity in LCDD is related to renal complications, including hypertension, nephrotic syndrome, and progression to end-stage renal disease (ESRD). Liver dysfunction can also occur, with progression to hepatic failure. Other symptoms of LCDD relate to congestive heart failure, peripheral neuropathy, and skin lesions secondary to the deposition of light chains.
Prognosis
Median survival for patients with light-chain deposition disease (LCDD) is about 4 years. The largest series published so far has reported after a median follow-up of 27 months; 57% of patients developed uremia and 59% of patients died. [10] The prognostic factors are age, associated multiple myeloma, and extrarenal light-chain deposition. [5, 20] The presence of end-stage renal disease (ESRD) and need for dialysis did not change the outcome for these patients.
Overall survival has been shown to be influenced by patient age, the presence of coexisting multiple myeloma or cast nephropathy, and any evidence of extrarenal light-chain deposition.
The natural or treated history of LCDD is difficult to determine, as this disease is uncommon.