Background
In basic terms, lactic acid is essentially a carbohydrate within cellular metabolism and its levels rise with increased metabolism during exercise and with catecholamine stimulation. Glucose-6-phosphate is converted anaerobically to pyruvate via the Embden-Meyerhof pathway. Pyruvate is in equilibrium with lactate with a ratio of about 25 lactate to 1 pyruvate molecules. Thus, lactate is the normal endpoint of the anaerobic breakdown of glucose in the tissues. The lactate exits the cells and is transported to the liver, where it is oxidized back to pyruvate and ultimately converted to glucose via the Cori cycle. However, all tissues can use lactate as an energy source, as it can be converted quickly back to pyruvate and enter into the Krebs cycle. In the setting of decreased tissue oxygenation, pyruvate is not readily metabolized and its intracellular levels rise, causing lactate levels to rise proportionally. With a persistent oxygen debt and overwhelming of the body's buffering abilities (whether from long-term dysfunction or excessive production), hyperlacticaemia and metabolic acidosis ensue, commonly referred to as lactic acidosis. [1, 2] (See Etiology.)
Lactic acid exists in two optical isomeric forms, L-lactate and D-lactate.
L-lactate is the most commonly measured level, as it is the only form produced in human metabolism. Its excess represents increased anaerobic metabolism due to tissue hypoperfusion. (See Workup.)
D-lactate is a byproduct of bacterial metabolism and may accumulate in patients with short-gut syndrome or in those with a history of gastric bypass or small-bowel resection. [3]
By the turn of the 20th century, many physicians recognized that patients who are critically ill could exhibit metabolic acidosis unaccompanied by elevation of ketones or other measurable anions. In 1925, Clausen identified the accumulation of lactic acid in blood as a cause of acid-base disorder. Several decades later, Huckabee's seminal work firmly established that lactic acidosis frequently accompanies severe illnesses and that tissue hypoperfusion underlies the pathogenesis. In their classic 1976 monograph, Cohen and Woods classified the causes of lactic acidosis according to the presence or absence of adequate tissue oxygenation. (See Presentation and Differentials.)
The causes of lactic acidosis are listed in the chart below.
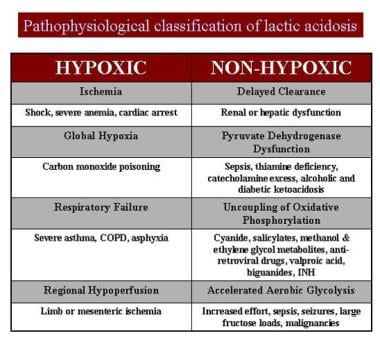 Pathophysiologic classification of lactic acidosis.
Pathophysiologic classification of lactic acidosis.
Go to Acute Lactic Acidosis for complete information on this topic.
Hyperlactatemia versus lactic acidosis
The normal blood lactate concentration in unstressed patients is 0.5-1 mmol/L. Patients with critical illness can be considered to have normal lactate concentrations of less than 2 mmol/L. Hyperlactatemia is defined as a persistent, mild to moderate (2-4 mmol/L) increase in blood lactate concentration without metabolic acidosis, whereas lactic acidosis is characterized by persistently increased blood lactate levels (usually >5 mmol/L) in association with metabolic acidosis.
Hyperlactatemia can occur in the setting of adequate tissue perfusion, intact buffering systems, and adequate tissue oxygenation.
Lactic acidosis, on the other hand, is associated with major metabolic dysregulation, tissue hypoperfusion, the effects of certain drugs or toxins, and congenital abnormalities in carbohydrate metabolism. It also occurs as a result on markedly increased transient metabolic demand (eg, postseizure lactic acidosis). Congenital lactic acidosis is secondary to inborn errors of metabolism, such as defects in gluconeogenesis, pyruvate dehydrogenase, the tricarboxylic acid (TCA) cycle, or the respiratory chain. These disorders generally reflect situations in which the disposal of pyruvate by biosynthetic or oxidative routes is impaired.
Lactic acidosis may not necessarily produce acidemia in a patient. The development of lactic acidosis depends on the magnitude of hyperlactatemia, the buffering capacity of the body, and the coexistence of other conditions that produce tachypnea and alkalosis (eg, liver disease, sepsis). Thus, hyperlactatemia or lactic acidosis may be associated with acidemia, a normal pH, or alkalemia.
Numerous etiologies may be responsible for the presence of lactic acidosis, most commonly circulatory failure and hypoxia. Evidence suggests increased morbidity and mortality for patients with persistently elevated or increasing lactate levels. Identification and discontinuation of any offending agents and treatment of known pathology should occur promptly. Although treatment with buffering agents remains controversial, their use should be considered in certain instances with the assistance of appropriate medical consultation. In addition, there is a growing body of literature showing the benefit of acute medical management, appropriate intervention (including early goal directed therapy) and lactate clearance.
Metabolic acidosis
Metabolic acidosis is defined as a state of decreased systemic pH resulting from either a primary increase in hydrogen ion (H+) or a reduction in bicarbonate (HCO3-) concentrations. [4] In the acute state, respiratory compensation of acidosis occurs by hyperventilation resulting in a relative reduction in PaCO2. Chronically, renal compensation occurs by means of reabsorption of HCO3. [5]
Acidosis arises from an increased production of acids, a loss of alkali, or a decreased renal excretion of acids. The underlying etiology of metabolic acidosis is classically categorized into those that cause an elevated anion gap and those that do not. Lactic acidosis, identified by a state of acidosis and an elevated plasma lactate concentration, is one type of anion gap metabolic acidosis and may result from numerous conditions. [6]
Severe metabolic acidosis with arterial pH of less than 7.2 is associated with impaired cardiac contractility and suboptimal response to exogenous catecholamines. Elevation of serum lactate concentration may have negative inotropic effects independent of serum pH.
Go to Metabolic Acidosis and Pediatric Metabolic Acidosis for complete information on these topics.
Types of lactic acidosis
Cohen and Woods divided lactic acidosis into 2 categories, type A and type B. [5, 7]
Type A is lactic acidosis occurring in association with clinical evidence of poor tissue perfusion or oxygenation of blood (eg, hypotension, cyanosis, cool and mottled extremities). It can be caused by the overproduction of lactate or the underutilization of lactate. In cases of overproduction, circulatory, pulmonary, and hemoglobin transfer disorders are commonly responsible.
In cases of underutilization of lactate, liver disease, gluconeogenesis inhibition, thiamine deficiency, and uncoupled oxidative phosphorylation can be responsible.
Type B is lactic acidosis occurring when no clinical evidence of poor tissue perfusion or oxygenation exists. However, in many cases of type B lactic acidosis, occult tissue hypoperfusion is now recognized to accompany the primary etiology.
Type B is divided into 3 subtypes based on underlying etiology.
Type B1 occurs in association with systemic disease, such as renal and hepatic failure, diabetes and malignancy. [8]
Type B2 is caused by several classes of drugs and toxins, including biguanides, alcohols, iron, isoniazid, zidovudine, and salicylates. [9]
Type B3 is due to inborn errors of metabolism.
Within these categories, septic shock may move from type A to type B, as the initial presentation is often associated with hypoperfusion, and with aggressive fluid resuscitation little evidence of tissue hypoperfusion exists, yet lactic acidosis often persists because of altered oxidative phosphorylation and leukocyte production of lactate caused by sustained increased inflammatory stimuli (see below).
Lactate production
The anaerobic metabolic pathway known as glycolysis is the first step of glucose metabolism and occurs in the cytoplasm of virtually all cells. The end product of this pathway is pyruvate, which can then diffuse into the mitochondria and be metabolized to carbon dioxide by another, more energy-efficient metabolic pathway, the Krebs cycle. The metabolism of glucose to pyruvate also results in the chemical reduction of the enzyme cofactor oxidized form nicotinic acid dehydrogenase (NAD+) to nicotinic acid dehydrogenase (NADH) (reduced form).
Erythrocytes are capable of carrying out glycolysis; however, these cells do not have mitochondria and cannot use oxygen to produce adenosine triphosphate (ATP). The pyruvate formed during glycolysis is metabolized by the enzyme lactate dehydrogenase to lactate. The anaerobic pathway is very inefficient, and only 2 moles of ATP are produced for each molecule of glucose that is converted to lactate. The lactate diffuses out of the cells and is converted to pyruvate and then is aerobically metabolized to carbon dioxide and ATP. The heart, liver, and kidneys use lactate in this manner.
Alternatively, hepatic and renal tissues can use lactate to produce glucose via another pathway referred to as gluconeogenesis. The metabolism of glucose to lactate by one tissue, such as red blood cells, and conversion of lactate to glucose by another tissue, such as the liver, is termed the Cori cycle.
Lactate is cleared from blood, primarily by the liver, with the kidneys (10-20%) and skeletal muscles doing so to a lesser degree. The ability of the liver to consume lactate is concentration-dependent and progressively decreases as the level of blood lactate increases. Lactate uptake by the liver also is impaired by several other factors, including acidosis, hypoperfusion, and hypoxia.
Cardiopulmonary failure, sepsis, trauma, thiamine deficiency, side effects of drugs and toxins, oncologic pathology, and various acquired and congenital diseases can lead to lactic acidosis. [1, 10, 11, 12]
Metabolic aspects of lactate production
The arterial concentration of lactate depends on the rates of its production and use by various organs. Blood lactate concentration normally is maintained below 2 mmol/L, although lactate turnover in healthy, resting humans is approximately 1300 mmol every 24 hours. Lactate producers are skeletal muscle, the brain, the gut, and the erythrocytes. Lactate metabolizers are the liver, the kidneys, and the heart. When lactate blood levels exceed 4 mmol/L, the skeletal muscle becomes a net consumer of lactate.
As mentioned above, lactate is a byproduct of glycolysis; it is formed in the cytosol catalyzed by the enzyme lactate dehydrogenase, as shown below:
Pyruvate + NADH + H+ = lactate + NAD+
This is a reversible reaction that favors lactate synthesis with the lactate-to-pyruvate ratio that is normally at 25:1. Lactate synthesis increases when the rate of pyruvate formation in the cytosol exceeds its rate of use by the mitochondria. This occurs when a rapid increase in metabolic rate occurs or when oxygen delivery to the mitochondria declines, such as in tissue hypoxia. Lactate synthesis also may occur when the rate of glucose metabolism exceeds the oxidative capacity of the mitochondria, as observed with administration of catecholamines or errors of metabolism.
Cellular energy metabolism and lactate production
Cells require a continuous supply of energy for protein synthesis. This energy is stored in the phosphate bonds of the ATP molecule. The hydrolysis of ATP results in the following reaction, where ADP is adenosine diphosphate and Pi is inorganic phosphate:
ATP = ADP + Pi + H+ + energy
With an adequate supply of oxygen, the cells use ADP, Pi, and H+ in the mitochondria to reconstitute ATP. During cellular hypoxia, the hydrolysis of ATP leads to accumulation of H and Pi in the cytosol. Therefore, ATP hydrolysis is the source of cellular acidosis during hypoxia and not the formation of lactate from glucose, which neither consumes nor generates H+. The glycolytic process may be viewed as the following:
D glucose + 2 ADP + 2 Pi = 2 lactate + 2 ATP
The hydrolysis of 2 ATP molecules formed from the metabolism of glucose produces H+, ADP, and Pi, as follows:
2 ATP = 2 ADP + 2 Pi + 2 H+ + energy
If the oxygen supply is adequate, the metabolites of ATP are recycled in the mitochondria and the cytosolic lactate concentration rises without acidosis. On the other hand, with cellular hypoxia, the equation of anaerobic glycolysis becomes the following:
D glucose = 2 lactate + 2 H+ + energy
A second cellular source of anaerobic ATP is the adenylate kinase reaction, also called the myokinase reaction, where 2 molecules of ADP join to form ATP and adenosine monophosphate (AMP).
ADP = AMP + Pi + H+ + energy
This reaction leads to increased intracellular levels of AMP, Pi, and H+. Thus, H+ is able to increase during hypoxemia without the notable increase in cellular lactate concentration.
Cellular transport of lactate
Intracellular accumulation of lactate creates a concentration gradient favoring its release from the cell. Lactate leaves the cell in exchange for a hydroxyl anion (OH-), a membrane-associated, pH-dependent, antiport system. The source of extracellular OH- is the dissociation of water into OH- and H+. Extracellular H+ combines with lactate leaving the cell, forming lactic acid, while intracellular OH- binds to H+ generated during the hydrolysis of ATP to form water. Therefore, cellular transport of lactate helps to moderate increases in cytosolic H+ resulting from hydrolysis of anaerobically generated ATP.
Etiology
Declines in cellular oxygen delivery lead to more oxygen extraction from the capillary blood. This action redistributes the cardiac output to organs according to their ability to recruit capillaries and also decreases the distance from the capillaries to the cells. With severe decreases in oxygen transport, compensatory increase in the oxygen extraction ratio is insufficient to sustain aerobic metabolism. Therefore, the cell must employ anaerobic sources of energy to produce ATP, resulting in the generation of lactate and H+.
The most frequent cause of lactic acidosis is poor tissue perfusion, which is induced by various shock states causing tissue hypoxia. In ischemic tissues of the skeletal muscle (and, less significantly, the intestine, erythrocytes, and brain), production of lactate is accelerated with a concomitant fall in lactate consumption by the liver, kidney, and myocardium. The accumulation of a normally balanced level of serum lactate overwhelms the body's buffering capacity and results in acidosis. [13, 14]
Lactate acidosis as a metabolic monitor of shock
Shock currently is conceptualized as a clinical syndrome resulting from an imbalance between tissue oxygen demands and tissue oxygen supply. Impaired oxygen delivery is the primary problem in hypovolemic, cardiogenic, distributive (septic), and obstructive (pericardial tamponade, tension pneumothorax) forms of shock. When tissue hypoxia is present, pyruvate oxidation decreases, lactate production increases, and ATP formation continues via glycolysis. The amount of lactate produced is believed to correlate with the total oxygen debt, the magnitude of hypoperfusion, and the severity of shock. Serial lactate determinations may be helpful in patients resuscitated from shock to assess the adequacy of therapies.
Hyperlactemia and lactic acidosis in sepsis
Patients who develop severe sepsis or septic shock commonly demonstrate hyperlactemia and lactic acidosis. The pathophysiology of sepsis associated lactic acidosis has not been well understood. Increased lactate production during anaerobic and aerobic metabolism and decreased lactate clearance are likely contributors to hyperlactemia. Patients with septic shock have lactate levels of more than 5 mmol/L, a lactate-to-pyruvate ratio greater than 10-15:1, and arterial pH of less than 7.35.
Following resuscitation from septic shock, some patients continue to demonstrate hyperlactemia (lactate 2-5 mmol/L), whereas blood pH is normal or alkalemic. These patients manifest increased oxygen consumption, insulin resistance, urea nitrogen excretion in urine, and a normal lactate-to-pyruvate ratio. Hyperlactemia likely occurs from increased production of pyruvate and equilibration with lactate, this has been termed "stress hyperlactemia." [15]
The mechanism of lactic acidosis in septic shock is continuing to be debated. Several studies have shown an elevated lactate-to-pyruvate ratio in septic shock, suggesting tissue hypoxia as the cause of lactic acidosis. However, other investigators have documented hyperlactemia in the absence of hypoxia.
The additional possible mechanisms for hyperlactemia include activation of glycolysis and inhibition of pyruvate dehydrogenase. Some investigators have observed that patients with sepsis have decreased lactate clearance rather than increased lactate production. Skeletal muscle and lung tissue have been shown to produce lactate during sepsis. Therefore, hyperlactemia may be secondary to increased lactate production in the gut, liver, lungs, and skeletal muscles; decreased lactate clearance in the liver; or a combination of both. Still, other investigators have suggested that hyperlactemia may occur secondary to down-regulating of pyruvate dehydrogenase in skeletal muscles by inflammatory mediators, rather than tissue hypoxia. [16]
Hyperlactemia in patients with sepsis is a marker of the severity of stress response. Hyperlactemia may possibly develop as a byproduct of overall acceleration in glycolysis in severe sepsis. This may well be an adaptive host mechanism designed to provide for efficient generation of energy in response to severe stress.
Limitations of lactic acidosis as a monitor of tissue perfusion
The use of lactate as an index of tissue perfusion has several limitations. The presence of liver disease causes a decreased ability to clear lactate during periods of increased production. Various causes of type B lactate acidosis may produce hyperlactemia and lactate acidosis in the absence of inadequate tissue perfusion. For significant increase in blood lactate to occur, lactate must be released into the systemic circulation and the rate of production must exceed hepatic, renal, and skeletal muscle uptake. Therefore, regional hypoperfusion of tissues may be present despite normal blood lactate concentrations.
Lactic acid levels can also lag several hours after the oxygen delivery critical threshold (DO2 crit) has been crossed. Indeed, patients may be accruing a significant amount of oxygen debt before lactate levels start to increase. It has been demonstrated that mixed venous saturation can fall below 50% before serum hyperlactatemia is evident. [17]
Lactic acidosis in disease
Lactic acidosis occurring from associated, underlying diseases, known as type B1 lactic acidosis, has been identified with diabetes mellitus, bowel ischemia, severe iron-deficiency anemia, liver disease, alcoholic ketoacidosis, pancreatitis, malignancy (leukemia, lymphoma, lung cancer), infection, renal failure, seizures, heat stroke, pheochromocytoma, thiamine deficiency, short gut syndrome, and other carbohydrate malabsorption syndromes. Type B3 lactic acidosis may result in persons with inborn errors of metabolism. These include glucose-6-phosphatase deficiency (von Gierke disease), fructose-1,6-diphosphatase deficiency, pyruvate carboxylase deficiency, pyruvate dehydrogenase deficiency, oxidative phosphorylation deficiency, and methylmalonic aciduria.
Lactic acidosis rarely may present in the MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and strokelike episodes), which appears to be caused by a point mutation in mitochondrial DNA tRNALeu (UUR) gene. This syndrome is characterized by migrainelike headaches, dementia, hearing loss, ataxia, and episodic vomiting
Aberrant lactate metabolism is frequently encountered among critically ill patients. Those with predisposing underlying disease states and medications portend an increased occurrence. The overall incidence of lactic acidosis in critically ill patients is unknown; however, increasing acid-base evaluations of critically ill patients indicate that its persistence increases associated morbidity and mortality. [1]
Medicines and toxins in lactic acidosis
Medicinal and toxic causes of lactic acidosis, specifically, type B2 lactic acidosis, include the following:
-
Acetaminophen
-
Alcohols and glycols (ethanol, ethylene glycol, methanol, propylene glycol)
-
Antiretroviral nucleoside analogs (zidovudine, didanosine, lamivudine)
-
Beta-adrenergic agents (epinephrine, ritodrine, terbutaline)
-
Biguanides (phenformin, metformin)
-
Cocaine
-
Cyanogenic compounds (cyanide, nitroprusside, amygdalin)
-
Diethyl ether
-
5-Fluorouracil
-
Halothane
-
Iron
-
Isoniazid
-
Propofol
-
Sugars and sugar alcohols (fructose, sorbitol, and xylitol)
-
Salicylates
-
Strychnine
-
Sulfasalazine
-
Valproic acid
A 2010 study by Salpeter et al found that the oral antihyperglycemic agent metformin, despite concerns to the contrary, is not associated with an increased risk for lactic acidosis compared with other antihyperglycemic treatments. [18]
Epidemiology
Prevalence of lactic acidosis is not known and is difficult to investigate; however, abnormal lactate metabolism is frequently encountered in patients who are critically ill.
Symptomatic hyperlactatemia is associated with antiretroviral therapy. In a large cohort of adults infected with the human immunodeficiency virus (HIV), hyperlactatemia was diagnosed in 64 patients. Incidences were 18.3 per 1000 person-years with antiretroviral therapy and 35.8 per 1000 person-years for stavudine (d4T) regimens.
Prognosis
Although the etiology of shock influences the probability of survival, blood lactate concentration has prognostic value. [19] Serum lactate levels of greater than 2.5 mmol/L have been associated with an increase in mortality rate.
Antiretroviral-associated hyperlactemia rarely causes death, but generally, the outcome for patients has been favorable after antiretroviral therapy has been stopped and supportive treatment with vitamins and antioxidants has been initiated.
Early diagnosis, vigilance, and routine measurements of the anion gap are crucial.
The clinical significance of mild hyperlactatemia greater than 3 mmol/L but less than 5 mmol/L is uncertain.
Mortality and morbidity
Patients who have an arterial lactate level of more than 5 mmol/L and a pH of less than 7.35 are critically ill and have a very poor prognosis. Multicenter trials have shown a mortality rate of 75% in these patients.
In another study, the median survival for patients with lactic acidosis and shock was 28 hours. Of these patients, 56% survived 24 hours and only 17% of the patients were discharged from the hospital. Nearly half of these patients showed evidence of multiorgan failure, and survival also correlated with the level of systolic blood pressure. Patients with a systolic blood pressure of less than 90 mm Hg had a 12.5% survival rate, while patients with a systolic pressure of more than 90 mm Hg had a 55% survival rate at 72 hours.
In an observational study of intensive care patients, the mortality rate was highest for patients with lactic acidosis (56%), compared with anion gap acidosis (39%). A stepwise logistic regression model identified serum lactate, anion gap acidosis, phosphate, and age as independent predictors of mortality. Overall, patients with metabolic acidosis were nearly twice as likely to die as patients without metabolic acidosis. [11]
In post–cardiac arrest patients who are comatose after return of spontaneous circulation, a greater percent decrease in lactate over the first 12 hours is associated with better survival and neurologic outcome. Additionally, those patients who did not survive or had a poor neurologic outcome had higher lactate levels at 0, 12, and 24 hours post cardiac arrest. [20]
In the context of sepsis, the Surviving Sepsis Campaign guidelines recommend the use of early goal-directed therapy for patients with a serum lactate level greater than 4.0 mmol/L. [21] Historically, the clinical significance of an intermediately elevated lactate level was unknown. However, a recent systematic review found that in emergency department patients with suspected sepsis, lactate levels between 2.0 and 3.9 mmol/L were associated with a moderate-to-high risk of mortality, even in patients without hypotension. [22]
More recent research has suggested that in patients in the emergency department with sepsis, early lactate normalization within the first 6 hours of resuscitation was a strong independent predictor of survival. [23] To date, no randomized, controlled studies have addressed whether early lactate normalization improves outcomes.
Patients exhibiting a disorder of lactate metabolism are typically significantly ill and are at risk for developing multiple organ failure. Patients suffer a hospital mortality rate that increases nearly linearly with the concentration of serum lactate. Several studies have shown that vigilant correction of hyperlactemia is associated with decreased morbidity and mortality. The mortality rate of patients with a serum lactate level greater than 2 mmol/L persisting after 24 hours with an associated acidemia approaches 70%. [24]
-
Pathophysiologic classification of lactic acidosis.





