Practice Essentials
Alpha2–plasmin inhibitor (alpha2-PI), also known as alpha2-antiplasmin, is a protein that is primarily synthesized by the liver and is present in plasma and platelets. It is the most important physiologic inhibitor of plasmin, and thus plays a major role in regulating blood coagulation. [1] Congenital alpha2-PI deficiency, due to homozygous carriage of a defective SERPINF2 gene, which encodes alpha2-PI, is an extremely rare cause of a bleeding disorder. Most of those individuals experience prolonged bleeding and bruising following minor trauma and bleeding into the joints, similar to the manifestations seen in patients with hemophilia. [2] Acquired alpha2-PI deficiency may occur in patients with severe liver disease
To date, fewer than 20 cases of congenital homozygous alpha 2-PI deficiency and 8 molecular defects of the alpha2-PI gene have been reported worldwide. [3, 4, 5] The first reported case involved a 25-year-old Japanese homozygous male born of consanguineous parents. [6] He had a lifelong history of severe bleeding, starting with bleeding from the umbilical cord at birth. The patient experienced hematomas, prolonged bleeding from cuts and after dental extraction, and muscle and joint bleeds following minor trauma. [6] Central nervous system (CNS) bleeding has also been described in a Dutch patient who was homozygously deficient. [7]
In 3 homozygous patients (sisters) from another Japanese family, bleeding was milder, with umbilical bleeding at birth followed by hematomas, gingival bleeding, and epistaxis without joint bleeding. The levels of alpha2-PI were undetectable in all of the patients.
Most reported heterozygous patients did not have clinically significant bleeding, although some had a bleeding disorder. Currently, the reasons for variability in bleeding manifestations in heterozygous persons with alpha2-PI deficiency are unclear.
In addition to routine coagulation studies, the workup for patients with possible alpha2-PI deficiency includes antigenic and functional alpha2-PI assays, which are performed in specialized laboratories (see Workup). Treatment depends on the severity of a bleeding episode. Minor bleeding can be handled with oral antifibrinolytic drugs (eg, epsilon-aminocaproic acid tranexamic acid), but more extensive bleeding may require temporary supplementation with fresh-frozen plasma or solvent/detergent-treated plasma. Bleeding into critical sites may also require surgical intervention. Prophylactic use of oral antifibrinolytics may be indicated for patients undergoing major surgery, or for long-term prevention in with inherited alpha2-PI deficiency who experience severe bleeding. (See Treatment and Medication.)
Pathophysiology
Alpha2–plasmin inhibitor (alpha2-PI) is the most important physiologic inhibitor of plasmin, which is the principal protease of the fibrinolytic pathway. Plasminogen activators convert the zymogen plasminogen to the active enzyme plasmin, which then hydrolyzes susceptible arginine and lysine bonds in a variety of proteins. [8, 9, 10]
Plasmin has a broad range of actions. Plasmin not only degrades fibrin, which is its principal substrate, but it also degrades fibrinogen, factors V and VIII, proteins involved in platelet adhesion (glycoprotein I and vWF), platelet aggregation (glycoprotein IIb/IIIa) and maintenance of platelet aggregates (thrombospondin, fibronectin, histidine-rich glycoprotein), and the attachment of platelets and fibrin to the endothelial surface.
A positive feedback mechanism exists whereby plasmin acts to further increase the generation of plasmin by converting Glu-plasminogen to Lys-plasminogen; Lys-plasminogen is more susceptible to activation by plasminogen activators. In addition, other noncoagulation proteins, such as complement, growth hormone, corticotropin, and glucagon, are substrates for plasmin. Therefore, the reasons for the bleeding disorder that develops due to the actions of excess unfettered and unneutralized plasmin are easily comprehended.
Alpha2-PI belongs to the serpin family of inhibitors, is synthesized by the liver, and is present in plasma as a single-chain protein at approximately half the concentration of plasminogen. Two forms of alpha2-PI are present in blood; 70% of alpha2-PI binds plasminogen and has inhibitory activity, whereas the remaining 30% is in a nonbinding form. The nonbinding form is a degradation product of the binding form and has little inhibitory activity.
A small amount of alpha2-PI present in platelets contributes to inhibition of fibrinolysis in platelet-containing thrombi. Activated factor XIII (FXIIIa) cross-links alpha2-PI to the a-chains of fibrin(ogen), thus making a cross-linked fibrin clot more resistant to lysis by plasmin. [11, 12, 13]
Alpha2-PI reacts very rapidly with plasmin to form a stable plasmin-inhibitor complex. This interaction is central to the physiologic control of fibrinolysis and irreversibly inhibits plasmin activity, which in turn, partially degrades alpha2-PI. The plasmin–alpha2-PI complex is cleared more rapidly from the circulation. The half-life of the complex is approximately 12 hours compared with the longer half-life of 3 days for native alpha2-PI.
Alpha2-PI performs several functions. It inhibits free plasmin rapidly and more readily than fibrin-bound plasmin. It is cross-linked to fibrin, thus conferring resistance to degradation by plasmin, and it interferes with the adsorption of plasminogen to fibrin. As a result, recent clots are more susceptible than older clots to degradation by plasmin.
Several other proteins are also involved in the complex process of regulation of fibrinolysis in vivo. Physiologically, the end result is that the hemostatic plug (fibrin and platelet clot) is protected from premature breakdown, leaving the fibrin meshwork intact so that it functions not only in hemostasis but also in wound repair as a scaffold for regenerating cells.
As the principal inhibitor of plasmin, alpha2-PI plays a key role in the physiologic control of fibrinolysis by helping localize reactions to the sites where they are needed and by helping prevent systemic spillover. When the amount of plasmin generated exceeds the capacity of alpha2-PI to neutralize plasmin (since, in plasma, plasminogen levels are twice those of alpha2-PI) alpha2-macroglobulin can function as a less efficient backup inhibitor. Note the image below.
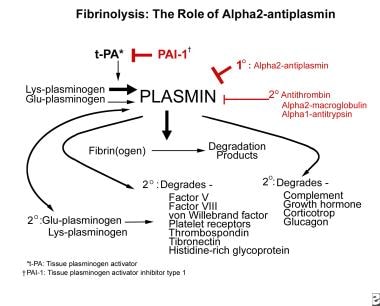 The role of alpha2-plasmin inhibitor (alpha2-antiplasmin) in fibrinolysis.
The role of alpha2-plasmin inhibitor (alpha2-antiplasmin) in fibrinolysis.
Conceptually, alpha2-PI neutralizes plasmin at various sites of plasmin production, including in the fibrin clot, on the surface of cells, and in the fluid phase (For an excellent diagram showing these details, see Figure 2 in Castellino FJ, Ploplis VA. Plasminogen and streptokinase. In: Bachmann F, ed. Fibrinolytics and Antifibrinolytics. Berlin: Springer-Verlag; 2001:26-56.) [14]
Other inhibitors, such as antithrombin, alpha1-antitrypsin, and C1 inactivator of complement, have in vitro antiplasmin activity, but these inhibitors may play only a minimal role in vivo.
In the absence of alpha2-PI, plasmin degrades the primary hemostatic platelet-fibrin plug, thereby interfering with adequate primary hemostasis. Although fibrin formation is unimpaired, subsequent accelerated lysis of the formed fibrin plug (fibrinolysis) leads to the onset of delayed bleeding.
In pathologic states, in which there is an endogenous excessive activation of plasminogen or a secondary infusion of activators, such as tissue plasminogen activator (tPA) and streptokinase, sudden generation of large amounts of plasmin overwhelms the neutralizing capacity of alpha2-PI. In addition to degrading the primary fibrin-platelet plug, excess plasmin degrades circulating fibrinogen (fibrinogenolysis) and factors V and VIII, adding to the hemorrhagic diathesis.
Most patients with an inherited homozygous alpha2-PI have a clinically significant bleeding disorder that is characterized by prolonged bleeding and bruising following minor trauma and bleeding into the joints, similar to the manifestations seen in patients with hemophilia. [2] Gene knockout mouse models of alpha2-PI deficiency show the expected accelerated clot lysis, but the mice do not manifest the bleeding disorder that is seen in humans. [15]
Epidemiology
Frequency
United States
Very few cases of inherited alpha 2-plasmin inhibitor (alpha 2-PI) deficiency have been reported; therefore, data do not exist to determine the true frequency. In the next several years, as widespread high-throughput genomic testing becomes commonplace, the frequency of genetic defects will be known, and the frequency of these rare disorders can then be determined.
The frequency of acquired alpha 2-plasmin inhibitor deficiency depends on the frequency of the underlying disorders. As discussed in Causes, excessive bleeding can occur when alpha 2-PI levels are deficient.
Mortality/Morbidity
Homozygous patients with alpha 2-plasmin inhibitor deficiency have severe bleeding that requires plasma therapy to limit the bleeding and to maintain plasma levels until the acute bleeding resolves.
Recurrent joint bleeds can lead to acute and chronic arthropathy, as occurs in severe hemophilia. Appropriate physical therapy, joint replacement, and treatment of chronic debilitating viral illnesses, such as hepatitis and acquired immunodeficiency syndrome (AIDS) and its sequelae, are needed in patients with alpha 2-plasmin inhibitor deficiency. Death may occur due to a CNS bleed or after major trauma. Note the following:
-
Patients with an inherited homozygous alpha 2-plasmin inhibitor deficiency have a clinically significant bleeding disorder characterized by easy bruising, delayed onset of bleeding following trauma or surgery, menorrhagia, epistaxis, hematuria, and bleeding into joints, similar to the manifestations seen in patients with hemophilia.
-
The frequency of a bleeding disorder reportedly varies among patients who are heterozygous for alpha 2-PI deficiency and is characterized by a milder bleeding disorder in most heterozygotes, with a tendency to worsen with age.
-
The bleeding in patients with acquired disorders associated with alpha 2-plasmin inhibitor deficiency is described in Causes.
Race-, sex-, and age-related demographics
Note the following:
-
No ethnic predilection for alpha 2-plasmin inhibitor deficiency is known at this time because the overall number of reported cases is so small.
-
Alpha 2-plasmin inhibitor deficiency is inherited as an autosomal recessive trait. Males and females are equally susceptible.
-
Clinical manifestations of alpha 2-plasmin inhibitor deficiency may start at birth, with excess bleeding from the umbilical cord. Bleeding manifestations may start later in childhood, when trauma and minor cuts occur with increasing activity. Menorrhagia manifests following puberty in women.
-
The role of alpha2-plasmin inhibitor (alpha2-antiplasmin) in fibrinolysis.