Overview of Platelet Disorders
The hemostatic system consists of platelets, coagulation factors, and the endothelial cells lining the blood vessels. The platelets arise from the fragmentation of the cytoplasm of megakaryocytes in the bone marrow and circulate in blood as disc-shaped anucleate particles for 7-10 days.
Under physiological circumstances, the resistance of the endothelial cell lining to interactions with platelets and coagulation factors prevents thrombosis. When endothelial continuity is disrupted and the underlying matrix is exposed, a coordinated series of events are set in motion to seal the defect (primary hemostasis). See the image below.
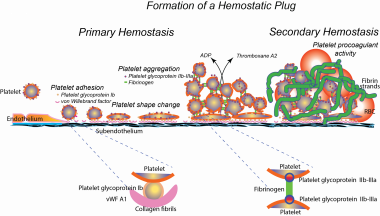 Normal hemostasis. ADP = adenosine diphosphate; RBC = red blood cell; vWF = von Willebrand factor
Normal hemostasis. ADP = adenosine diphosphate; RBC = red blood cell; vWF = von Willebrand factor
Platelets play a primary role in this process, interacting with subendothelium-bound von Willebrand factor (vWf) via the membrane glycoprotein (GP) Ib complex. This initial interaction (platelet adhesion) sets the stage for other adhesive reactions that allow the platelets to interact with other agonists in the vicinity of vessel injury, such as adenosine 5'-diphosphate (ADP), subendothelial collagen, and thrombin. These interactions further activate platelets.
The platelet GP IIb/IIIa complex mediates platelet-to-platelet interactions (platelet aggregation). On resting platelets, GP IIb/IIIa is unable to bind fibrinogen or vWf. Platelet activation allows binding of these proteins, which bridges adjacent platelets. Morphologically, the platelets change dramatically from discs to spiny spheres in a process called shape change.
Platelets contain two unique types of granules: alpha granules and dense granules. The alpha granules contain hemostatic proteins such as fibrinogen, vWf, and growth factors (eg, platelet-derived growth factor and transforming growth factors). The dense granules contain proaggregatory factors such as ADP, calcium, and 5-hydroxytryptamine (serotonin). During activation, the granules are centralized and their contents are discharged into the lumen of the open canalicular system, from which they are then released to the exterior (the release reaction).
Once activated, platelets have two major mechanisms to recruit additional platelets to the growing hemostatic plug. They release proaggregatory materials (eg, ADP) by the release reaction, and they synthesize thromboxane A2 from arachidonic acid. Thus, the release reaction and prostaglandin synthesis act to consolidate the initial hemostatic plug by promoting the participation of other platelets in the growing hemostatic plug. See the image below.
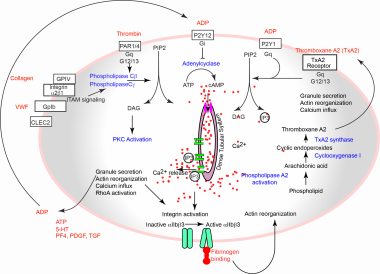 Platelet activation pathways. 5-HT = 5-hydroxytryptamine; ADP = adenosine diphosphate; ATP = adenosine triphosphate; cAMP = cyclic adenosine monophosphate; CLEC2 = C-type lectin-like receptor 2; DAG = diacylglycerol; GP = glycoprotein; IP3 = inositol trisphosphate; ITAM = immunoreceptor tyrosine-based activation motif; P2Y = purinergic receptor; PAR = protease-activated receptor; PDGF = platelet-derived growth factor; PF4 = platelet factor 4; PIP2 = phosphatidylinositol 4,5-bisphosphate; PKC = protein kinase C; TGF = transforming growth factor; VWF= von Willebrand factor
Platelet activation pathways. 5-HT = 5-hydroxytryptamine; ADP = adenosine diphosphate; ATP = adenosine triphosphate; cAMP = cyclic adenosine monophosphate; CLEC2 = C-type lectin-like receptor 2; DAG = diacylglycerol; GP = glycoprotein; IP3 = inositol trisphosphate; ITAM = immunoreceptor tyrosine-based activation motif; P2Y = purinergic receptor; PAR = protease-activated receptor; PDGF = platelet-derived growth factor; PF4 = platelet factor 4; PIP2 = phosphatidylinositol 4,5-bisphosphate; PKC = protein kinase C; TGF = transforming growth factor; VWF= von Willebrand factor
In addition, when platelets are activated, negatively charged phospholipids move from the inner to the outer leaflet of the membrane bilayer. This negative surface provides binding sites for enzymes and cofactors of the coagulation system, resulting in the formation of a clot (secondary hemostasis).
Pathophysiology of Platelet Disorders
Platelet disorders lead to defects in primary hemostasis and produce signs and symptoms different from coagulation factor deficiencies (disorders of secondary hemostasis). The body's reaction to vessel wall injury is rapid adhesion of platelets to the subendothelium. The initial hemostatic plug, composed primarily of platelets, is stabilized further by a fibrin mesh generated in secondary hemostasis. The arrest of bleeding in a superficial wound, such as the bleeding time wound, almost exclusively results from the primary hemostatic plug. See the image below.
Hence, primary hemostatic disorders are characterized by prolonged bleeding time, and the characteristic physical examination findings are petechiae and purpura. In comparison, defects in secondary hemostasis result in delayed deep bleeding (eg, into muscles and joints) and the characteristic physical examination finding is hemarthrosis. Hemarthrosis and muscle hematomas are not present in primary hemostatic disorders.
Autoimmune Thrombocytopenias
Immune thrombocytopenia
Immune thrombocytopenia (ITP) is one of the most common autoimmune disorders. ITP is caused by autoantibodies to platelets. The antigenic target in most patients appears to be the platelet GP IIb/IIIa complex. Platelets with antibodies on their surface are trapped in the spleen, where they are efficiently removed by splenic macrophages.
The mechanism of origin of these antibodies is not known. These antibodies may be directed toward viral antigens and then cross-react with platelet antigens. They persist because of the failure of immune surveillance mechanisms to repress these autoantibodies.
These antibodies can also react with the developing megakaryocytes in the bone marrow, leading to decreased production of platelets (ineffective thrombopoiesis). The success of thrombopoietin agonist therapy in chronic ITP underscores this mechanism as a major factor in inducing thrombocytopenia.
An alternative theory has been proposed by Malik et al after identifying cytotoxic CD8+ T cells as an antibody-independent mechanism of platelet destruction in chronic ITP. These researchers demonstrated that adults with chronic ITP have clonal expansion of terminally differentiated effector memory CD8+ T cells (TEMRA), compared with age-matched controls. TEMRAs form aggregates with platelets, release interferon gamma, and trigger platelet activation and apoptosis via the T-cell receptor–mediated release of cytotoxic granules. [1]
ITP occurs commonly in otherwise healthy individuals and only rarely as the initial manifestation of lupus and other autoimmune disorders. Human immunodeficiency virus (HIV) infection is often associated with ITP in both adults and children. A female predominance is most pronounced among middle-aged patients. In patients over age 60 years, however, the incidence of ITP increases with age with no difference between the sexes. [2]
ITP occurs in two distinct clinical types: (1) an acute self-limiting form observed almost exclusively in children (five cases per 100,000 persons) and (2) a chronic form, observed mostly in adults (three to five cases per 100,000 persons) and rarely in children. [3, 4, 5]
Acute ITP
Acute ITP affects males and females equally and has a peak incidence in children aged 3-5 years. Most patients have a history of an antecedent acute viral syndrome.
The onset is sudden, with symptoms and signs depending on the platelet count. Bleeding is usually mild, unless the platelet count drops below 20,000/µL. With platelet counts from 20,000/µL to 50,000/µL, petechiae and ecchymoses are observed following mild trauma. With platelet counts less than 10,000/µL, generalized petechiae, ecchymoses, and mucosal bleeding occur. With platelet counts below 2000/µL, widespread ecchymoses, hemorrhagic bullae, and retinal hemorrhage occur.
Physical examination reveals only the presence of petechiae and ecchymoses. The presence of lymphadenopathy or splenomegaly suggests other secondary causes of thrombocytopenia rather than ITP.
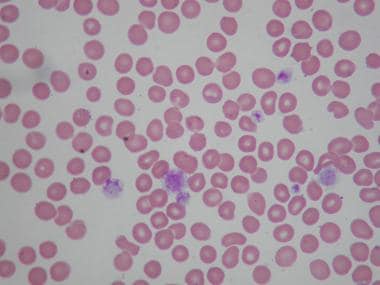
The peripheral smear shows a decreased number of platelets. Often, the smear shows giant platelets, which is a reflection of the increased megakaryocytic mass in the marrow induced by thrombopoietin stimulation (see the images below). Because of their size, these platelets are not counted as platelets in most particle counters.
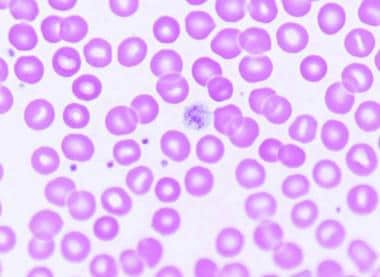 Examination of peripheral smears in immune thrombocytopenia often shows giant platelets. These platelets reflect the increased megakaryocytic mass in the marrow.
Examination of peripheral smears in immune thrombocytopenia often shows giant platelets. These platelets reflect the increased megakaryocytic mass in the marrow.
At times, the smear may show eosinophilia and lymphocytosis, possibly reflecting hypersensitivity to the inciting viral antigens. The bone marrow shows an increase in the number of megakaryocytes and signs of thrombopoietin-induced megakaryocyte stimulation (increase in number and ploidy, decrease in cytoplasm; see image below). This is the source for large platelets in the periphery.
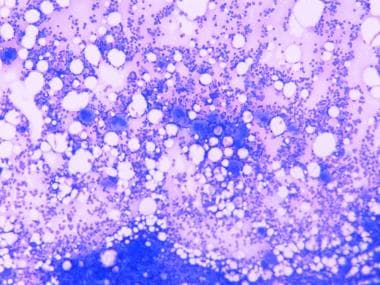 Bone marrow in immune thrombocytopenia reveals an increased number of megakaryocytes.
Bone marrow in immune thrombocytopenia reveals an increased number of megakaryocytes.
Thrombocytopenia in an otherwise healthy child with normal white and red blood cell counts almost always results from ITP. Findings from a careful history and physical examination help exclude other causes of thrombocytopenia, such as lupus and HIV infection.
Acute leukemia is unlikely to manifest as an isolated thrombocytopenia without any abnormalities in the smear. Bone marrow examination is necessary only if atypical features (ie, other abnormalities in the smear, sternal tenderness, lymphadenopathy, splenomegaly) or an unusual clinical course is evident.
Chronic ITP
This condition is typically observed in adults aged 20-40 years. It has an insidious onset, and a history of an antecedent infection need not be present. Unlike childhood ITP, chronic ITP is more common in females than in males. As in childhood ITP, the bleeding manifestations depend on the platelet count. [6, 7, 8]
The diagnosis of ITP is established by the exclusion of other causes of thrombocythemia. The peripheral blood film should be examined to rule out thrombotic thrombocytopenic purpura (TTP) (fragments) or spurious thrombocytopenia resulting from clumping or platelet satellitism (see image below).
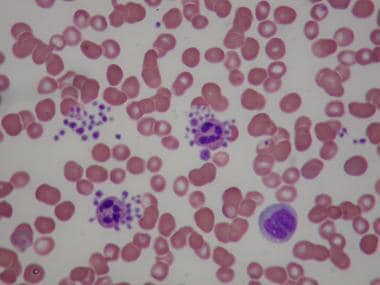 Spurious thrombocytopenia. Peripheral smear from a patient reported to have platelet counts ranging from 10,000 to 150,000/μL on various occasions. The smear shows clumping of the platelets and satellitism involving neutrophils and platelets.
Spurious thrombocytopenia. Peripheral smear from a patient reported to have platelet counts ranging from 10,000 to 150,000/μL on various occasions. The smear shows clumping of the platelets and satellitism involving neutrophils and platelets.
Often, the smear shows giant platelets, which is a reflection of the increased thrombopoietin-induced stimulation of bone marrow. Bone marrow examination, which is not always necessary, shows increased megakaryocytes.
Thrombocytopenia in pregnancy
Gestational thrombocytopenia is defined as mild thrombocytopenia in an otherwise healthy pregnancy. How this can be distinguished from a mild form of ITP is not clear. This disorder does not result in neonatal thrombocytopenia.
Hypertensive disorders of pregnancy (ie, preeclampsia/eclampsia syndrome) are associated with increased platelet turnover, even when the platelet count is normal. Controlling hypertension and delivering the fetus lead to restoration of the platelet count.
Occasionally, thrombocytopenia is associated with hemolysis and elevated liver enzymes (ie, hemolysis, elevated liver enzymes, and low platelet [HELLP] syndrome). This serious disorder often mimics thrombotic thrombocytopenic purpura.
Posttransfusion purpura
Platelet GP IIb/IIIa is a major antigen in platelets and is polymorphic. Most individuals have leucine at position 33; these antigens are designated as human platelet alloantigen (HPA)–1a or PLA1; thus, homozygotes are HPA-1a/HPA-1a. A small number of individuals, approximately 1-3% of random populations, have proline at position 33; this is designated as HPA-1b or PLA2. [9] Homozygotes with proline (HPA-1b/HPA-1b, or PLA2/PLA2) are termed PLA1-negative.
When HPA-1a–negative patients receive blood products from HPA-1a–positive individuals, they produce an antibody reactive against HPA-1a. This alloantibody destroys the transfused platelets and the patient's own platelets, leading to a severe form of thrombocytopenia that lasts for several weeks and, sometimes, several months. It is likely these alloantibodies induce autoantibody formation. Other platelet alloantigens are occasionally implicated in posttransfusion purpura.
Posttransfusion purpura typically occurs 10 days following a transfusion. This syndrome can be induced by a small amount of platelets contaminating a red blood cell transfusion or, occasionally, following fresh frozen plasma (FFP) transfusion. The thrombocytopenia responds to intravenous immunoglobulin (IVIG).
A population-based study of inpatients aged 65 years and older found that posttransfusion purpura occurred at an overall rate of 1.8 per 100,000 hospital stays. Risk of posttransfusion purpura were significantly higher with platelet-containing transfusions, greater number of units transfused, and underlying health conditions including a history of cardiac arrhythmias, coagulopathy, leukemia, and transplantation. [10]
Thrombocytopenia and COVID-19
Thrombocytopenia is infrequently seen in mild or asymptomatic cases of COVID-19. Of patients with moderate to severe COVID-19, 5-40% develop thrombocytopenia. A meta-analysis suggested an association between thrombocytopenia at admission and increased severity of COVID-19. Multiple mechanisms are involved in the pathogenesis of COVID-19–related thrombocytopenia, including bone marrow suppression, consumption in microthrombi in the lung, and destruction by autoantibodies and immune complexes. [11]
Vaccine-induced immune thrombotic thrombocytopenia
Vaccine-induced immune thrombotic thrombocytopenia (VITT) is a new syndrome reported in individuals receiving certain COVID-19 vaccines. The syndrome consists of a consumptive coagulopathy, with thrombocytopenia, hypofibrinogenemia, and an elevated D-dimer concentration, typically developing after the first dose of vaccine. The pathophysiology of VITT is similar to that of heparin-induced thrombocytopenia, which is caused by platelet-activating antibodies against platelet factor 4.
Cerebral venous sinus thrombosis with thrombocytopenia and antiplatelet factor 4 (anti-PF4) antibodies were first reported with the AstraZeneca–Oxford vaccine. Subsequently both AstraZeneca–Oxford and Johnson & Johnson adenoviral vaccines were implicated. VITT is not seen with mRNA-based vaccines. [12]
VITT is a very rare complication; absolute risk is estimated to be below 5 per million vaccinated individuals. Because of its rarity, it is difficult to give any recommendation regarding treatment. It is reasonable to avoid heparin and platelet transfusions, and an expert panel recommends early use of intravenous immunoglobulin (1000 mg/kg daily for 2 days). [13]
Neonatal alloimmune thrombocytopenia
The prevalence of neonatal alloimmune thrombocytopenia is approximately one case in 200 term pregnancies; for clinically apparent disease, the prevalence is one case in 1500 term pregnancies. It is the most common cause of severe neonatal thrombocytopenia. [14]
This disorder occurs when maternal antibodies against fetal platelet antigens inherited from the father but absent in the mother cross the placenta and induce severe thrombocytopenia. Most cases of neonatal alloimmune thrombocytopenia are due to platelet antigens HPA-1a observed in mothers who are HPA-1b.
Less commonly, other platelet antigens, such as HPA-5b, are responsible for neonatal alloimmune thrombocytopenia. Thus, the pathophysiology of this disease is similar to that of the hemolytic disease of newborns. Unlike hemolytic disease, however, thrombocytopenia occurs during the first pregnancy in 50% of cases.
Typically, the diagnosis of neonatal alloimmune thrombocytopenia is considered when bleeding or severe thrombocytopenia occurs in a newborn after an otherwise uncomplicated pregnancy. The affected infant may have intracranial hemorrhage, and the disorder is associated with a relatively high mortality rate. The platelet count should be checked immediately after delivery and 24 hours later as it continues to fall.
Drug-induced thrombocytopenia
Drugs can induce thrombocytopenia by a number of mechanisms. [15] For example, cytotoxic drugs can inhibit platelet production in the bone marrow, as can thiazide diuretics, interferon, and alcohol.
More commonly, drug-induced thrombocytopenia results from the immunologic destruction of platelets. Drugs can induce antibodies to platelets, either acting as a hapten or as an innocent bystander. In addition, drugs such as gold salts and interferon can induce an ITP-like disorder.
Common drugs associated with thrombocytopenia include the following:
-
Amiodarone
-
Captopril
-
Sulfonamides
-
Glyburide (glibenclamide)
-
Carbamazepine
-
Ibuprofen
-
Cimetidine
-
Tamoxifen
-
Ranitidine
-
Phenytoin
-
Vancomycin
-
Piperacillin
The diagnosis of drug-induced thrombocytopenia is often empirical. A temporal relationship must be present between the initiation of the drug and the development of thrombocytopenia, with no other explanations for the thrombocytopenia. Recurrent thrombocytopenia following reexposure to the drug confirms the drug as the cause of thrombocytopenia.
Identifying the drug that is causing severe thrombocytopenia in an acutely ill patient who is taking multiple drugs is often challenging. A complete list of all available reports of drug-induced thrombocytopenia is available at Platelets on the Web.
Heparin causes a unique situation among drug-induced thrombocytopenias in that the antibodies also activate platelets and induce a hypercoagulable state. [16] See Heparin-Induced Thrombocytopenia.
Thrombotic thrombocytopenic purpura
TTP is a rare but serious disorder that was initially described as having a pentad of manifestations, as follows:
- Thrombocytopenia (with purpura)
- Red blood cell fragmentation
- Kidney failure
- Neurologic dysfunction
- Fever
Evidence indicates that TTP results from the abnormal presence of unusually large multimers of von Willebrand protein. [17] These ultralarge precursors, normally synthesized in the endothelial cells, are thought to induce the aggregation of platelets, causing platelet consumption. Occlusion of microvasculature by the platelets in the brain, kidney, and other organs leads to myriad symptoms.
Normally, these large multimers are processed to normal-sized multimers by ADAMTS13 (A Disintegrinlike And Metalloprotease with ThromboSpondin type 1 motif 13), a metalloproteinase plasma enzyme synthesized in the liver. [17] The sporadic forms of TTP are caused by an antibody or toxin inhibiting the activity of ADAMTS13. The chronic, recurrent form of TTP may result from a congenital deficiency of the enzyme. A TTP-like syndrome has been associated with lupus, pregnancy, HIV infection, and certain drugs (eg, quinine, ticlopidine, clopidogrel, cyclosporine, chemotherapeutic agents such as gemcitabine and mitomycin).
In addition to immune and congenital TTP, a third form of TTP has been tentatively identified, and termed unidentified TTP. In contrast to immune TTP, anti-ADAMTS13 IgG antibodies are lacking in unidentified TPP. Significantly, whereas in patients with immune TTP, ADAMTS13 circulates in plasma in an open configuration, which makes it available for autoantibodies to bind with it, in unidentified TPP ADAMTS13 circulates in closed conformation, as is typical of healthy persons. Compared with immune TTP, unidentified TTP is less likely to occur in women and tends to occur in older individuals; patients more often have associated cancers and less often have accompanying autoimmune diseases. [18, 19]
Patients with TTP often report an episode of flulike illness 2-3 weeks before presentation. Most patients with TTP do not have the classic pentad. The most common presentation is petechiae and neurologic symptoms that can range from headache and confusion to seizures and coma. Fever is present in slightly more than 50% of patients. Plasmapheresis is often started with a presumptive diagnosis of TTP in patients with a microangiopathic hemolytic picture.
For more information, see Thrombotic Thrombocytopenic Purpura.
Hemolytic-uremic syndrome
Patients with hemolytic-uremic syndrome (HUS) have vascular lesions indistinguishable from those observed in patients with TTP, but the renal vasculature exhibits the most lesions. Neurologic dysfunction is minimal.
HUS is a catastrophic illness that predominantly affects children aged 4-12 months, sometimes affects older children, and rarely affects adults. HUS follows an upper respiratory tract infection.
In the tropics, epidemics of HUS are frequent and resemble an infectious disease; however, no causative organism has been identified. In North America, Shigella-like toxins (secreted by Escherichia coli serotype 0157:H7 or Shigella dysenteriae serotype I) cause many cases of HUS. Diarrhea and abdominal cramps are very prominent symptoms. [17]
Disorders of Platelet Function
Functional disorders of platelets are relatively rare, and most of these disorders are mild. Thus, they may not be recognized early in life. [20]
von Willebrand disease
von Willebrand disease (vWD) is the most common inherited bleeding disorder. It is autosomal dominant, and its prevalence is estimated to be as high as one case per 1000 population.
The hallmark of von Willebrand disease is defective platelet adhesion to subendothelial components, caused by a deficiency of the plasma protein von Willebrand factor (vWF). This factor is a large, multimeric glycoprotein that is synthesized, processed, and stored in the Weibel-Palade bodies of the endothelial cells, and secreted constitutively and following stimulation.
vWF has a major role in primary hemostasis as mediator of the initial shear-stress–induced interaction of the platelet to the subendothelium via the GP Ib complex. In addition, vWF acts as a carrier and stabilizer of coagulation factor VIII by forming a complex in the circulation.
In the absence of vWF, the factor VIII activity level is low. Unlike classic hemophilia A, in which the factor VIII activity level is low because of a defect in factor VIII itself, in von Willebrand disease the factor VIII activity level is low because of a deficiency in its carrier protein.
von Willebrand disease is a relatively mild bleeding disorder, except in the occasional patient who is homozygous for the defect (type III); those patiens have severe bleeding often indistinguishable from classic hemophilia. In most cases, the bleeding manifestations are predominantly skin-related and mucocutaneous (eg, easy bruising, epistaxis, gastrointestinal hemorrhage).
Most bleeding episodes occur following trauma or surgery. In women, menorrhagia is common, often exacerbated by the concurrent use of nonsteroidal anti-inflammatory drugs.
Pregnant women with this disease usually do not have problems. However, vWF and factor VIII activity fall after childbirth. In a prospective observational cohort study of 32 women with von Willebrand disease and 40 women without von Willebrand disease, vWF levels peaked at 250% of baseline at 4 hours postpartum in the von Willebrand disease group and at 12 hours postpartum in the women without von Willebrand disease and then decreased rapidly, reaching baseline at 3 weeks in both groups.
Fifteen of the women with von Willebrand disease received treatment with desmopressin or vWF concentrate before or after delivery. Except immediately postpartum, treatment did not raise vWF and factor VIII levels to normal or to the levels seen in women with milder, untreated von Willebrand disease. These researchers concluded that even with treatment, women with vWD may be at increased risk for postpartum hemorrhage. [21]
Bleeding time is prolonged in persons with von Willebrand disease. However, the von Willebrand protein is phase-reactant (ie, synthesis increases in the presence of inflammation, infection, tissue injury, and pregnancy), which can result in normalization of levels in patients whose bleeding time would otherwise be mildly prolonged. This can complicate diagnosis.
In addition to the prolonged bleeding time, characteristic abnormalities in platelet aggregation tests occur. In patients with von Willebrand disease, platelets aggregate normally to all agonists except the antibiotic ristocetin, which induces binding of the von Willebrand protein to platelets, similar to what happens with platelets following vessel wall injury in vivo. Ristocetin-induced platelet aggregation correlates with the platelet-aggregating activity of the von Willebrand protein. The levels of coagulation factor VIII are also low, due to the decrease in vWf, the carrier of factor VIII in plasma.
Variants of von Willebrand disease
Although the common form of von Willebrand disease (type I) results from a quantitative deficiency of vWF, the variants result from qualitative abnormalities in the von Willebrand protein.
A common variant (type IIA) of von Willebrand disease results from functionally defective vWf that is unable to form multimers or be more susceptible to cleavage by ADAMTS13. Larger multimers are more active in mediating platelet vessel-wall interaction. In these variants, the factor VIII level may be normal.
In the type IIB variant, the von Willebrand protein has heightened interaction with platelets, even in the absence of stimulation. Platelets internalize these multimers, leading to a deficiency of von Willebrand protein in the plasma.
The type IIN (Normandy variant) is caused by a defect in the ability of vWF to bind with coagulation factor VIII, resulting in the shortened half-life of factor VIII in the plasma. The ristocetin-induced platelet aggregation and vWF antigens are normal. Previous descriptions of autosomal recessive hemophilia A are most likely von Willebrand disease type IIN.
Type IIM von Willebrand disease is due to a defect in binding to platelet glycoprotein Ib but no defect in multimerization. In this variant, the ristocetin cofactor activity and ristocetin-induced platelet aggregation are decreased but the vWF antigen and multimers are present in normal levels.
A disorder of platelet GP Ib, mimicking type IIB von Willebrand disease, has also been described. In this condition, increased affinity for von Willebrand protein in the resting stage leads to the deletion of plasma von Willebrand protein. This disease is called pseudo von Willebrand disease or platelet-type von Willebrand disease.
Type III von Willebrand disease is a severe form that is characterized by very low levels of vWf and clinical features similar to hemophilia A, but with autosomal recessive inheritance. This condition results from a homozygous state or double heterozygosity.
Bernard-Soulier syndrome
Bernard-Soulier syndrome results from a deficiency of platelet glycoprotein protein Ib, which mediates the initial interaction of platelets with the subendothelial components via the von Willebrand protein. It is a rare but severe bleeding disorder. Platelets do not aggregate to ristocetin. The platelet count is low, but, characteristically, the platelets are large, often the size of red blood cells, and may be missed on complete blood counts because most automatic counters do not count them as platelets. [22] See the image below.
 Peripheral smear from a patient with Bernard-Soulier syndrome showing giant platelets. These platelets are not counted as platelets in most particle counters.
Peripheral smear from a patient with Bernard-Soulier syndrome showing giant platelets. These platelets are not counted as platelets in most particle counters.
Glanzmann thrombasthenia
Glanzmann thrombasthenia results from a deficiency of the GP IIb/IIIa complex. Platelets do not aggregate to any agents except ristocetin. The more severe type I results from a complete absence of the GP IIb/IIIa complex, whereas in the milder type II, some of the GP IIb/IIIa complex is retained.
Both Bernard-Soulier syndrome and Glanzmann thrombasthenia are characterized by lifelong bleeding. Although platelet transfusions are effective, they should be used only for severe bleeding and emergencies, because alloantibodies often develop in these patients.
Disorders of Secretion and Thromboxane Synthesis
During primary hemostasis, thromboxane synthesis and released ADP play a major role. A mild bleeding diathesis ensues if these mechanisms are deficient. Thromboxane synthesis disorders are almost always caused by aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs).
Because aspirin irreversibly inactivates cyclooxygenase in platelets, its effect lasts throughout the lifespan of platelets, which is approximately 1 week. Approximately 10% of new nonaspirinized platelets are produced daily; after 3 days have elapsed since the discontinuation of aspirin (30% nonaspirinized platelets available), the bleeding time normalizes.
Other NSAIDs are competitive inhibitors of cyclooxygenase, and their effect on platelets depends on the half-life of the drug. For example, the effect of ibuprofen, and most other NSAIDs, lasts only 1 day.
Mutations in the enzyme that converts arachidonic acid to thromboxane A2 have been described and are associated with a lifelong bleeding diathesis. Similarly, an absent or defective receptor for thromboxane A2 also leads to an aspirin-like aggregation defect.
In disorders of release reaction, platelets fail to secrete proaggregatory ADP following activation. The defects result from either the absence of granules in platelets or the defective storage of ADP. Inherited deficiency of ADP receptor P2Y12, characterized by mild bleeding diathesis, has also been described.
ADP is present in the dense granules of platelets as a storage pool, which is not used in the normal metabolic activity of platelets (in contrast to the metabolic pool). These disorders are often associated with other systemic abnormalities (eg, Hermansky-Pudlak syndrome).
Disorders of secretion and thromboxane synthesis are mild platelet disorders and often respond to desmopressin (DDAVP) infusion, which seems to improve hemostatic function. If severe bleeding is present, these disorders can also be managed effectively with platelet transfusions. Platelet transfusions, however, should be avoided as much as possible because they can induce alloantibodies. Cryoprecipitate has also been reported to be very effective.
Platelet Dysfunction in Uremia
Abnormal bleeding is common in patients with uremia. Bleeding time is generally very prolonged. The bleeding has the characteristics of a platelet disorder, and gastrointestinal tract bleeding is the most frequent manifestation.
Platelet function in uremic patients improves after dialysis. A number of dialyzable factors have been shown to inhibit platelet function. Furthermore, uremic platelets synthesize less thromboxane A2, and the blood vessels in patients with uremia produce greater quantities of platelet-inhibitory prostaglandin.
Nitric oxide produced by the endothelial cells inhibits platelet function. Because the prolonged bleeding time and the hemostatic abnormalities are partly corrected by red blood cell transfusion or erythropoietin therapy, the failure of hemoglobin to quench excess nitric oxide synthesis has been suggested as partly responsible for the platelet dysfunction.
Epidemiology of Platelet Disorders
Inherited hemostatic disorders are relatively rare. The prevalence of von Willebrand disease has been estimated at 1 case per 1000-5000 population.
In contrast, acquired hemostatic disorders are common, and ITP is one of the most common autoimmune disorders. The acute self-limiting form of ITP, which is observed almost exclusively in children, occurs at a rate of 5 cases per 100,000 population, and the chronic form, which is observed mostly in adults occurs at a rate of 3-5 cases per 100,000 population.
Unlike hemophilia, most inherited disorders of platelets are not X-linked, and they are equally distributed in both sexes. Acute ITP is also observed equally in both sexes. Chronic autoimmune thrombocytopenia is more common in females than in males.
Clinical Presentation of Platelet Disorders
Patient history
History and physical examination findings help clinicians to distinguish between primary and secondary hemostatic disorders and to determine whether the disorder is inherited or acquired.
Epistaxis is common in individuals with primary hemostatic disorders, but it is also common in healthy individuals. Details about the frequency, duration, packing requirement, and previous treatment (cautery or transfusion) are helpful for assessing the severity of bleeding (see also Anterior Epistaxis Nasal Pack and Posterior Epistaxis Nasal Pack).
Bleeding gums is a common sign in persons with primary disorders of hemostasis. The bleeding could be spontaneous or it could be associated with brushing or flossing. Bleeding from tooth extractions is possible. A molar tooth extraction is a traumatic procedure; uneventful extraction of a molar is unlikely in a patient with a severe bleeding disorder.
Hemoptysis, hematemesis, hematuria, hematochezia, and melena are rarely the initial symptoms of a bleeding disorder. However, these may be exacerbated by an underlying bleeding disorder.
Menstrual history is important. Metromenorrhagia is often observed in women with primary hemostatic disorders. This is especially common in those with von Willebrand disease and is often exacerbated by the NSAIDs used to treat dysmenorrhea. Bleeding after childbirth may be the first manifestation of a mild bleeding disorder.
Bleeding in the joints is the hallmark of hemophilia and other secondary hemostatic disorders. Details of previous surgeries, including the amount of blood transfused, if any, are helpful.
In males, excessive bleeding following circumcision is often the initial manifestation of a congenital bleeding disorder. Delayed bleeding from the umbilical stump is characteristic of a factor XIII deficiency.
Defective wound healing is observed in individuals with a factor XIII deficiency and abnormal fibrinogens. Medication history findings may be helpful because aspirin often accentuates a preexisting bleeding disorder. A history of previous iron therapy for anemia may be useful.
Physical examination
Bruising is common in individuals with a platelet disorder. A careful physical examination often reveals signs of a hemostatic disorder (see image below).
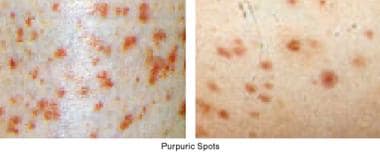
Petechiae are pinpoint hemorrhages (< 2 mm) in the skin, and purpura (0.2-1 cm) and ecchymoses are larger hemorrhages. The purpura is not palpable, in contrast to the palpable and sometimes tender purpura observed in patients with vasculitis.
Initially, purpura tends to form in the areas of increased venous pressure, such as the legs. Petechiae and purpura may develop following the application of a sphygmomanometer cuff.
Splenomegaly is not observed in the typical patient with ITP. The spleen can engulf platelets and be several times normal size without becoming palpably enlarged.
Hemarthrosis and deep muscle hematomas are unusual in patients with primary hemostatic disorders.
Etiology of Platelet Disorders
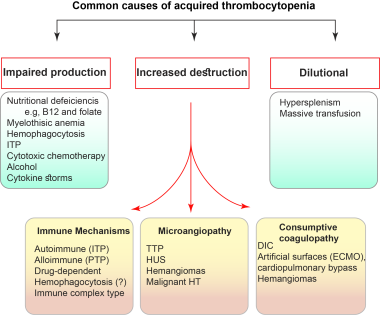
Platelet disorders can involve either a decreased number of platelets (thrombocytopenia) or defective platelet function. Functional disorders of platelets can be inherited (rare) or acquired (common). Platelet aggregation tests are useful in differentiating various disorders of platelet function. In all cases of thrombocytopenia, the peripheral blood smear must be reviewed to confirm the thrombocytopenia. This review is crucial.
Spurious thrombocytopenia can occur due to aggregates forming in the specimen. In addition, dilutional thrombocytopenia may occur in situations of fluid replacement or blood component replacement without platelet support.
Thrombocytopenia can be further divided into increased destruction or decreased production. Thrombocytopenia resulting from increased destruction occurs either by an immune mechanism or increased consumption.
Platelets are consumed intravascularly by the activation of the coagulation process (diffuse/disseminated intravascular coagulation [DIC]) or by deposition on damaged endothelial cells (microangiopathy). Production defects result from those diseases that cause bone marrow failure, such as aplastic anemia, infiltration by leukemia or another malignancy, fibrosis or granulomatous disorders, or tuberculosis.
Causes of thrombocytopenia related to increased destruction include (1) immune thrombocytopenias (eg, autoimmune, alloimmune, drug-induced) and (2) increased consumption (eg, DIC, TTP).
Causes of thrombocytopenia related to decreased production include bone marrow depression and inherited disorders. Genetic defects have been defined for 30 forms of inherited thrombocytopenia, but the underlying genetic or molecular mechanisms remain unidentified for nearly 50% of cases. [23]
Inherited disorders of platelet function are as follows:
-
Disorders of platelet adhesion (von Willebrand disease, Bernard-Soulier syndrome)
-
Disorders of aggregation (Glanzmann thrombasthenia)
-
Disorders of secretion
-
Disorders of thromboxane synthesis
Acquired disorders of platelet function may result from the following:
-
Drugs (eg, aspirin, other NSAIDs, alcohol)
-
Uremia
-
Paraproteins
-
Fibrin degradation products
Laboratory Studies
A variety of studies are available for the assessment of patients with possible platelet disorders. A complete blood count and peripheral blood smear are the key laboratory components of the diagnosis of immune thrombocytopenia (ITP) and thrombotic thrombocytopenic purpura (TTP). In pediatric patients, immunoglobulin assays are often performed to exclude common variable immune deficiency (CVID) as a cause of ITP. [24]
Bleeding assessment tools
Structured bleeding assessment tools (BATs) such as from the International Society on Thrombosis and Haemostasis (ISTH‐BAT) may be utilized as a screening instrument in specific patients and implemented in the diagnostic evaluation. [25, 26, 27] The ISTH‐BAT records both the presence and the severity of bleeding at 14 important sites. Although it has been validated in patients with suspected von Willebrand disease (vWD), the diagnostic utility of the ISTH‐BAT with regard to platelet function disorders requires more study. [26]
ISTH-BAT scores were calculated in a study of 61 patients with suspected hereditary/congenital platelet disorders (HPDs). ISTH-BAT identified patients with suspected HPDs but lacked a robust association with laboratory abnormalities. The researchers reported that 19 patients had thrombocytopenia and 46 had positive ISTH-BAT scores. Additionally, 24 had platelet transmission electron microscopy (PTEM) abnormalities (10 dense granule deficiency, 14 other ultrastructural abnormalities). Positive ISTH-BAT scores were associated with thrombocytopenia (P < 0.0001) and abnormal PTEM results (P = 0.002). [27]
The utility of the ISTH‐BAT was studied in a large cohort of patients with a well‐defined diagnosis of inherited platelet disorder in comparison with two parallel cohorts, one of patients with type 1 vWD (vWD‐1) and one of healthy controls. Of the 1098 participants, 196 had inherited platelet function disorders (IPFD) and 286 had inherited platelet number disorders (IT). The IPFD subjects had significantly higher bleeding score (BS) (median 9) than vWD‐1 patients (median 5), a higher number of hemorrhagic symptoms (4 vs 3) and higher percentage of patients with clinically relevant symptoms (score > 2).
Discrimination power of the ISTH‐BAT between IPFD and healthy controls was excellent (0.9 < area under the curve [AUC] < 1); between IPFD and vWD‐1 and IPFD and inherited thrombocytopenia, it was moderate (0.7 < AUC < 0.9). However, it proved inaccurate (AUC ≤0.7) in discriminating IT from healthy controls. [25]
Peripheral smear
Careful examination of the peripheral smear is essential in a patient with thrombocytopenia.
Spurious thrombocytopenia due to platelet clumping or platelets adhering to neutrophils (platelet satellitism) can be seen on a smear (see image below).
Spurious thrombocytopenia. Peripheral smear from a patient reported to have platelet counts ranging from 10,000 to 150,000/μL on various occasions. The smear shows clumping of the platelets and satellitism involving neutrophils and platelets.
Giant platelets are often seen in patients with ITP (see image below).
Examination of peripheral smears in immune thrombocytopenia often shows giant platelets. These platelets reflect the increased megakaryocytic mass in the marrow.
Rare disorders, such as Bernard-Soulier syndrome, can be diagnosed based on the results from the peripheral smear (see image below). Careful examination of the smear is essential to exclude TTP and rare instances of acute leukemia presenting as thrombocytopenia.
Peripheral smear from a patient with Bernard-Soulier syndrome showing giant platelets. These platelets are not counted as platelets in most particle counters.
In TTP, a striking degree of red blood cell fragmentation is seen in addition to thrombocytopenia (see image below).
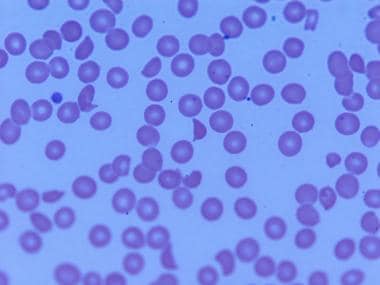 Peripheral smear in thrombotic thrombocytopenic purpura shows red blood cell fragments and basophilic cells, in addition to thrombocytopenia.
Peripheral smear in thrombotic thrombocytopenic purpura shows red blood cell fragments and basophilic cells, in addition to thrombocytopenia.
The minimum criteria for the diagnosis of TTP are thrombocytopenia and microangiopathic hemolytic anemia without an apparent etiology.
Examination of the smear shows thrombocytopenia and a microangiopathic picture (characteristic helmet cells/schistocytes and basophilic red blood cells) (see image below). In addition, the lactate dehydrogenase (LDH) level is high, with brisk reticulocytosis. Signs of intravascular coagulation are characteristically absent in patients with TTP.
Platelet-associated immunoglobulin G
The autoantibodies responsible for autoimmune thrombocytopenia do not induce complement-mediated lysis. Furthermore, when platelets are destroyed in the circulation, they internalize plasma proteins, including immunoglobulin. Platelets also have low affinity to the crystallizable fragment (Fc) receptor, FcgRIIa, that binds immunoglobulin.
In patients with autoimmune thrombocytopenia, the larger platelets have proportionately more membrane surface and more Fc receptor than normal platelets. For these reasons, the detection of increased platelet-associated immunoglobulin is not useful because it is elevated in almost all conditions associated with thrombocytopenia, which limits the value of this test in the diagnosis of ITP. Specialized tests to identify antibodies that react specifically against platelet membrane glycoproteins are not clinically available.
Bleeding time
This is a valuable test for disorders of primary hemostasis; however, this test is highly operator-dependent and is not recommended as a routine screening test. Primary hemostasis bleeding time is performed by measuring the duration required for bleeding to stop from a fresh superficial cut (1 mm deep, 1 cm long) made on the volar surface of the forearm using a template under standard conditions.
Under these conditions, the cessation of bleeding results from the formation of a primary hemostatic plug. A fairly linear correlation exists between bleeding time and platelet counts of 10,000-100,000/µL. Bleeding time is prolonged with platelet counts below 75,000/µL, although that finding provides no insight into why the count is low.
Primary hemostasis bleeding time should not be performed on patients with thrombocytopenia. A prolonged bleeding time with a normal platelet count is very significant and indicates a qualitative platelet disorder.
In disorders of secondary hemostasis (eg, hemophilia A and B), bleeding time is almost invariably normal.
In vitro platelet function analyzer 100
The platelet function analyzer 100 (PFA-100) is a bench-top automated instrument that assesses primary hemostasis under shear stress. The PFA-100 uses a disposable test cartridge that contains a membrane impregnated with collagen plus ADP (Col/ADP membrane) or epinephrine (Col/Epi membrane). A blood sample of 0.8 mL of citrated blood is placed in a cup and is aspirated through the aperture. The shear stress and the agonists in the membrane activate platelets, leading to platelet aggregation.
The end point, expressed as closure time, is when blood flow stops because of occlusion of the aperture by platelet aggregates.
The platelet aggregate formation depends on the following:
- vWf binding to collagen-coated nitrocellulose membranes
- Platelet adhesion to vWF via platelet GP Ib platelet activation
- Platelet aggregation mediated by the interaction of GP IIb/IIIa with vWF and fibrinogen
Normal closure times range from 77 to 133 seconds for the Col/ADP membrane and 98 to 185 seconds for the Col/Epi membrane. The closure time using the Col/Epi cartridge is abnormal in patients with congenital platelet function defects, von Willebrand disease, or aspirin ingestion, whereas the closure time with the Col/ADP cartridge is abnormal mainly in patients with von Willebrand disease or congenital disorders.
Aspirin prolongs the closure time 94% of the time with the Col/Epi cartridge and only 27% of the time with the Col/ADP cartridge. Glanzmann thrombasthenia, Bernard-Soulier syndrome, and most mild von Willebrand diseases are associated with a prolonged closure time with both cartridges, whereas a storage pool defect and giant platelet thrombopathy have a prolonged closure time only with the Col/Epi cartridge.
The advantages of this instrument include simplicity and reproducibility. The PFA-100 has been reported to have a coefficient of variation of less than 10%. It may be useful for determining global platelet function and for assessing the efficacy of antiplatelet therapy.
Platelet aggregation
Platelet aggregation is measured by turbidimetric methods. When platelets aggregate, the opalescent suspension of platelet-rich plasma becomes clearer and allows more light transmission. The extent of aggregation is determined by measuring the increase in light transmission. See the image below.
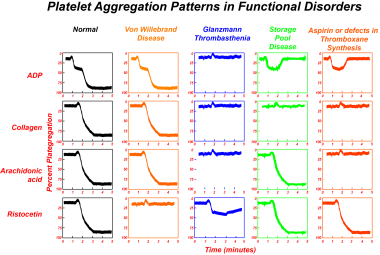
Small doses of ADP (< 1 µmol) induce a reversible form of platelet aggregation (primary wave), unaccompanied by thromboxane synthesis or release of intraplatelet ADP. However, with increasing doses of ADP, sufficient stimulation of platelets occurs and leads to the release of intraplatelet ADP and the synthesis of thromboxane A2 from arachidonic acid, thus resulting in more pronounced irreversible aggregation (secondary wave).
Ristocetin induces platelet aggregation by inducing von Willebrand protein binding to the platelet GP Ib complex.
Platelet aggregation tests are useful in distinguishing different disorders of platelet function. They are also particularly useful in the diagnosis of von Willebrand disease, in which ristocetin-induced platelet aggregation is defective.
Imaging Studies
Imaging studies are not necessary to diagnose uncomplicated ITPs. Rarely, platelet survival studies may be necessary to document decreased platelet survival before splenectomy in a patient with possible bone marrow hypofunction. Typically, the platelet half-life is decreased from the normal 5-7 days. A normal platelet survival curve is not consistent with increased splenic destruction.
In a patient whose condition has relapsed following splenectomy, an indium-labeled platelet imaging study is sometimes useful for localizing an accessory spleen.
Bone Marrow Examination
Bone marrow examination is not necessary in most cases of platelet disorders. The isolated presence of large platelets in the peripheral blood, in the absence of any other signs of bone marrow dysfunction, is very suggestive of normal marrow activity.
Bone marrow examination is necessary in patients whose condition has an atypical course, have splenomegaly, or will undergo splenectomy, and in patients over age 60 years, as thrombocytopenia may be the initial manifestation of myelodysplastic syndrome.
Bone marrow examination in patients with ITP shows megakaryocytic hyperplasia (see image below). Quantifying the megakaryocytes in the bone marrow is technically difficult. Usually, 2-3 megakaryocytes are present in each spicule in typical marrow. Clusters of immature megakaryocytes are often observed in patients with ITP.
Bone marrow in immune thrombocytopenia reveals an increased number of megakaryocytes.
Treatment of ITP in Children
Because acute immune thrombocytopenia (ITP) in children is self-limited, most physicians do not routinely treat it. Treatment is necessary only to prevent intracranial or other serious internal hemorrhage. [6] The rate of intracranial hemorrhage is very low, possibly less than 0.1%, and occurs with platelet counts of 10,000-20,000/µL.
Most physicians treat children with ITP when the platelet count is less than 20,000/µL. That is an arbitrary threshold, however, and current guidelines from the American Society of Hematology (ASH) recommend that children with no bleeding or mild bleeding (defined as bruising and petechiae, with no mucosal bleeding) be managed with observation alone regardless of platelet count. [28]
Treatment options include intravenous immune globulin (IVIG) and corticosteroids, alone or in combination. In select cases, anti-D immunoglobulin may be used.
IVIG (0.8-1 g/kg for 2 d) results in a prompt rise in the platelet count, and this response confirms the diagnosis of acute ITP. The mechanisms of action of IVIG are not clear. Suggested mechanisms include blocking the macrophage Fc receptors, suppressing autoantibody production by providing anti-idiotypes, and stimulating the clearance of autoantibodies.
The adverse effects of IVIG include fever, nausea, vomiting, and, occasionally, kidney failure. IVIG is also very expensive compared with prednisone and is not available in all countries.
Oral prednisone (4 mg/kg, with tapering and discontinuation by day 21) or IV methylprednisolone (30 mg/kg for 3 d) is also effective, although recovery is not as quick as with IVIG. The mode of action of prednisone is probably multifold, and includes the following:
-
Decreasing antibody production
-
Increasing platelet formation
-
Decreasing macrophage-mediated clearance of platelets in the spleen
-
Immunomodulating the immune response
The combination of steroids and IVIG is synergistic and can be used in patients with imminent hemorrhage.
Inducing a mild hemolytic state by administering anti-D immunoglobulin (25-50 μg/kg for 2 d) is effective in individuals who are Rh positive, but is recommended only in those who have a negative direct antiglobulin test (DAT) and who have not undergone splenectomy. This therapy is less expensive than IVIG. However, the US Food and Drug Administration (FDA) has provided a warning and specific monitoring requirements because cases of fatal intravascular hemolysis have been reported with anti-D immunoglobulin. The ASH guidelines acknowledge the black box warning related to fatal intravascular hemolysis, but note that these are rare events. [28]
With these modalities, the platelet counts in most children can be maintained at more than 30,000/µL until spontaneous remission occurs.
Other supportive measures include avoiding drugs that impair platelet function (eg, aspirin) and avoiding competitive contact sports.
ITP may develop in children after measles, mumps, and rubella (MMR) vaccination. However, such cases occur at a lower rate than after natural measles or rubella infection, and recurrence of ITP has not been reported after MMR vaccination of unimmunized patients with ITP or re-vaccination of patients with previous nonvaccine- or vaccine-associated ITP. [24]
ASH guidelines therefore recommend that unimmunized children with a history of ITP receive their first MMR vaccine on schedule. In a child with ITP who has already received the first dose of MMR vaccine and whose titers demonstrate full immunity, no further MMR vaccine should be given. If the child does not have adequate immunity, MMR vaccination should continue according to the recommended schedule. [24]
Treatment of Chronic ITP in Children
Approximately 25% of children with ITP do not undergo spontaneous remission within 6 months and have a chronic course with remissions and relapse similar to adult-onset chronic ITP. The rate of chronic ITP appears to increase with advancing age, rising from 23.1% in children younger than 12 months to 47.3% in children older than 10 years, in one study. [29]
In a retrospective study of 47 children with chronic ITP who were not receiving corticosteroid therapy, 21 (44.7%) showed spontaneous remission according to new International Working Group standards, maintaining a platelet count of at least 100×109/L approximately 3-4 years following their diagnosis; 31 children (66%) maintained a platelet count of 50×109/L or higher through 5.4 years of follow-up. Lower age at diagnosis and longer follow-up were significantly associated with a better prognosis. [30]
Because of the likelihood of spontaneous remission, splenectomy should be avoided if possible. Furthermore, splenectomy in patients younger than 6 years is associated with severe post-splenectomy sepsis. If treatment with corticosteroids, IVIG, or anti-D has been successful, these agents may be used prophylactically while waiting for a possible spontaneous remission.
Although a number of agents (eg, dapsone) have been studied for treatment of pediatric patients with chronic ITP that is refractory to conventional agents, the American Society of Hematology found insufficient evidence to support recommendations regarding their use. American Society of Hematology guidelines suggest the following for treatment of refractory chronic ITP in children and adolescents who have non–life-threatening mucosal bleeding and/or diminished health-related quality of life [28] :
-
Thrombopoietin receptor agonist (eltrombopag or romiplostim) over rituximab or splenectomy
-
Rituximab over splenectomy
Children with chronic ITP who are scheduled for splenectomy should receive pneumococcal and Haemophilus influenzae vaccines before the operation. Many physicians recommend that patients receive a prophylactic antibiotic regimen after splenectomy.
Alloimmune thrombocytopenia in neonates
If left untreated, alloimmune thrombocytopenia in neonates persists from a few days to up to 3 weeks. The treatment of choice is the administration of IVIG and maternally compatible platelets. Maternal platelets should be radiated to avoid graft versus host disease in the infant, and washed to reduce the antibody concentration. Response to steroids is rare. [14]
Treatment of Chronic ITP in Adults
No consensus has been reached regarding when to start steroid therapy for chronic immune thrombocytopenia (ITP) in adults and how long to treat it. [3, 7, 31, 32] The 2019 American Society of Hematology guidelines recommend against treatment of patients with a platelet count ≥30 × 109/L. For newly diagnosed patients with a platelet count < 30 × 109/L, guidelines suggest treatment with corticosterioids. For patients with a platelet count < 20 × 109/L, the guidelines suggest hospital admission for treatment. For first-line treatment, the guidelines further recommend a short course (≤6 weeks) of steroids over a prolonged course (> 6 weeks including treatment and taper) of steroids. Additional first-line treatment suggestions include the following [28] :
-
Either prednisone (0.5-2.0 mg/kg per day) or dexamethasone (40 mg per day for 4 days) as the type of corticosteroid
-
Corticosteroids alone rather than rituximab and corticosteroids
Corticosteroid therapy
A course of steroid therapy is often administered upon the initial diagnosis in an effort to induce a sustained remission. The treatment of choice is high-dose oral dexamethasone given at a dose of 40 mg daily for 4 consecutive days and then stopped. If the platelet count remains below 30 × 109/L or bleeding symptoms occur by day 10, a second 4-day course of dexamethasone (40 mg daily) can be given.
Wei et al reported that a high-dose dexamethasone regimen resulted in a higher overall initial response, higher complete response rates, and shorter time to response, compared with prednisone. [33] In another study, a 4-day course of high-dose dexamethasone (40 mg/d) was reported as an effective initial therapy for adults with ITP, with 50% of patients showing sustained platelet count of over 50,000/µL. [34]
Prednisone is usually administered at a dose of 1 mg/kg. Approximately two thirds of patients can be expected to show a therapeutic response with steroid therapy.
Several observational studies have shown that high-dose dexamethasone improved durable platelet count responses compared with standard-dose prednisone. However, a meta-analysis of randomized trials found that durable platelet count responses were similar with both regimens. [35] However, high-dose dexamethasone was associated with earlier improvement in platelet count, fewer bleeding events, and less toxicity than prednisone over the course of treatment. An additional advantage is the ease of administration of high-dose dexamethasone, as a course lasts only 4 days, as opposed to prednisone, which is often continued as a prolonged course with longer corticosteroid exposures.
Steroids are usually continued until the platelet count reaches normal or greater than 50,000/µL, and then they are gradually tapered in 4-6 weeks. Methylprednisolone (30 mg/kg IV days 1-3, tapered every third day to 1 mg/kg) has also been used with similar results.
In general, only 15-25% of patients with chronic ITP are expected to have a lasting remission; the remainder have disease characterized by frequent relapses and remissions.
Even if the platelet count normalizes, many patients can maintain platelet counts of more than 20,000-30,000/µL with lower doses of steroids during times of relapse. However, in approximately one third of patients with chronic ITP, steroids are not effective, either because of a failure in response or a steroid requirement that leads to unacceptable adverse effects (eg, glucose intolerance, GI bleeding).
Intravenous immunoglobulin
IVIG at a dose of 1 gm/kg can be used as initial therapy in addition to steroids when a rapid increase in platelet count is desired. IVIG can also be used as the initial treatment of choice in patients with HCV or HIV.
IVIG (1 g/kg/d for 1-2 d) induces a short-term increase in the platelet count, starting within several days and lasting approximately 2-3 weeks, both in patients who have undergone splenectomy and in those who have not. No clear evidence indicates that repeated infusions induce a lasting remission.
Anti-D immunoglobulin (WinRho, 50-75 μg/kg IV) is also as effective as IV immunoglobulin in Rh-positive adults with an intact spleen. Massive intravascular hemolysis with DIC and occasional death has occurred with the use of anti-D immunoglobulin.
Both IVIG and anti-D immunoglobulin are relatively expensive therapy for adults compared with steroids, and these agents are primarily used on an interim basis during a crisis (eg, before splenectomy or major surgery).
Rituximab
Rituximab, a monoclonal antibody directed against the lymphocyte antigen CD20, has been shown to induce lasting remission in refractory ITP. Rituximab is effective in patients requiring high doses of steroids and in patients who have unacceptable side effects of steroids. Rituximab is given in a 375-mg/m2 infusion weekly for 4 weeks. About half the patients achieve complete or partial response to rituximab and about 20% have a lasting, treatment-free response. A meta-analysis of adults suggests that surgery could be delayed and may be avoided altogether for patients who received rituximab earlier in the course of therapy. [36] In women and in patients within 2 years of diagnosis, rituximab combined with three cycles of high-dose dexamethasone induces a higher response rate (up to 70%) than rituximab alone, dexamethasone alone, or the combination with one cycle of dexamethasone. [37]
Adverse effects of rituximab include the following:
-
Prolonged hypogammaglobulinemia
-
Poor response to vaccines
-
Reactivation of hepatitis B
-
Opportunistic infection in immunosuppressed patients
Splenectomy
With the advent of novel treatments in last two decades the incidence of splenectomy has decreased. Splenectomy is no longer considered the second-line therapy for steroid failures; rather, it is generally reserved for patients in whom multiple medical therapies have failed. Nevertheless, splenectomy is still considered the only curative treatment for ITP, with the highest long-response rates reported to date. [31]
The appropriate time to perform splenectomy is controversial. Most physicians wait for a year before recommending splenectomy because sometimes ITP goes into spontaneous remission, especially in younger patients. Often, other clinical considerations (eg, coexistence of diabetes or peptic ulcer disease [PUD]) may influence the decision for earlier splenectomy.
Splenectomy is effective because it removes the major site of platelet destruction and the major source of antiplatelet antibody synthesis. Even if complete remission is not achieved, the platelet count will be higher after splenectomy. [32] Before splenectomy, patients should receive a pneumococcal vaccine.
Approximately 10-20% of patients who undergo splenectomy remain thrombocytopenic and continue to have a bleeding risk that requires continued treatment. Both steroid therapy and splenectomy are considered to have failed in these patients, and treatment of these patients is challenging. An accessory spleen should be excluded as the cause of treatment failure after splenectomy. [32]
Thrombopoietin receptor agonists
In a significant proportion of patients with ITP, ineffective platelet production from megakaryocytes plays a major role in causing thrombocytopenia. Stimulation of megakaryopoiesis by exogenous agents improves the platelet count. Three thrombopoietin receptor agonists have been approved for the treatment of chronic refractory ITP: eltrombopag, romiplostim, and avatrombopag.
Eltrombopag
Eltrombopag is an oral nonpeptide thrombopoietin receptor agonist that interacts with the transmembrane domain of the thrombopoietin receptor and induces megakaryocyte proliferation and differentiation. It has been shown to increase the platelet count in refractory ITP and in thrombocytopenia associated with hepatitis C–induced cirrhosis. It will most likely also be effective in thrombocytopenia due to other causes, by stimulating megakaryocytes.
Eltrombopag is approved for treatment of refractory chronic ITP in patients who are at increased risk of bleeding because of their platelet count and clinical condition. It has also proved useful as bridging therapy for surgery, to provide a temporary pre-procedure boost in platelet count for patients with ITP who do not normally require therapy. [38]
Eltrombopag is given in doses of 25-75 mg daily. High-calcium food or an antacid containing aluminum and magnesium were associated with significantly reduced absorption of eltrombopag The manufacturer recommends taking the drug at least 2 hours before or 4 hours after any medications or products containing polyvalent cations such as antacids, calcium-rich foods, and mineral supplements.
The adverse effects include hepatotoxicity, worsening of cataracts, and increased bone marrow reticulin fibers In the open-label EXTEND study, [9] which evaluated long-term safety and efficacy of eltrombopag with a follow-up to 8 years in 300 adult patients, overall, 85.8% achieved a response with a platelet count ≥50 × 109/L at least once in the absence of rescue and 52 achieved a continuous response of 25 weeks or longer. Adverse effects such as thrombosis, hepatobiliary dysfunction, and bone marrow fibrosis were infrequent.
Romiplostim
Romiplostim is a thrombopoietin receptor agonist that consists of the human immunoglobulin Fc region covalently linked to a peptide sequence that binds to and activates the thrombopoietin receptor. The peptide sequence has no homology with human thrombopoietin so that the possibility of a cross-reacting antibody is minimized. The Fc domain extends the half-life of the molecule in the circulation.
Weekly subcutaneous doses of 1-7 μg/kg romiplostim can increase the platelet count in chronic ITP. The adverse effects include bone marrow reticulin formation.
In 2 parallel trials that assessed the long-term administration of romiplostim in 63 splenectomized and 62 nonsplenectomized patients with ITP, both the splenectomized and nonsplenectomized patients achieved durable platelet counts over a longer period with romiplostim than with placebo; patients receiving romiplostim were also more likely to reduce or discontinue concurrent other ITP therapy compared with patients in the placebo groups. [39]
The responses to thrombopoietin receptor agonists take 10-15 days, and, hence, they are unlikely to replace steroids or intravenous immunoglobulins as initial therapy. Furthermore, relapses are common, necessitating long-term therapy. Thrombopoietin receptor agonists may help postpone or even prevent splenectomies. The advantages of thrombopoietin receptor agonist therapy should be weighed against the risk of marrow fibrosis seen in the limited long-term outcome data. There is also a theoretical possibility that these agents increase the risk of hematologic malignancies, as thrombopoietin receptor is present in hematopoietic stem cells. Currently, these agents are recommended for ITP patients whose conditions are refractory to previous treatments, including splenectomy.
A meta-analysis of 5 randomized placebo-controlled trials in children with persistent or chronic ITP concluded that the overall response rates and the incidence of adverse events were similar between romiplostim and eltrombopag. [40] Nevertheless, eltrombopag might have lower risk of overall bleeding than romiplostim. Furthermore, cost and drug compliance should also be considered in clinical decision making, as romiplostim is given subcutaneously while eltrombopag is an oral medication.
Avatrombopag
Avatrombopag is an orally administered thrombopoietin receptor that is approved for treatment of chronic ITP in adults who have had an insufficient response to a previous treatment, and for treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. Avatrombopag does not require food restriction and it has not been associated with liver toxicity or portal vein thrombosis in patients with chronic liver disease.
Immunosuppression
Limited benefit may be observed using immunosuppression with cytotoxic agents. Azathioprine (150 mg/d) or cyclophosphamide (50-100 mg/d) has been used with some success. These cytotoxic drugs can cause myelosuppression, alopecia, hemorrhagic cystitis (cyclophosphamide), sterility, and secondary malignancy. They are given for a minimum duration and are withdrawn as soon as remission is achieved. Blood counts must be monitored during therapy.
Vincristine infusion (0.02 mg/kg) with a maximum dose of 2 mg every week for 3 weeks has also been shown to induce remission. Refractory ITP has also been treated with the combination chemotherapy regimen used for low-grade non-Hodgkin lymphoma (6 cycles of cyclophosphamide, vincristine, and prednisone), with some success.
Helicobacter pylori eradication
Several studies reported improved platelet counts in Helicobacter pylori–positive patients, following standard H pylori eradication therapy. Platelet count response rates vary internationally (presumably reflecting geographic differences in H pylori strains), with higher response rates observed in Japan, Italy, and Brazil than in the United States and other European countries. [41] American Society of Hematology guidelines recommend against routine testing for H pylori in children with chronic ITP, but suggest considering screening for H pylori in adult patients with ITP, and administering eradication therapy those patients who are found to have H pylori infection. [24, 28]
Other treatments
A number of treatments have been proposed for patients in whom splenectomy and steroid therapy have failed. Most of these treatments are not based on placebo-controlled studies, and evaluating their efficacy in a disease associated with spontaneous remissions and relapse is difficult.
Fostamatinib, an oral tyrosine kinase inhibitor, has been shown to increase the platelet count had a overall response of 18% in randomized placebo-controlled trial. (PMID: 29696684).
The anabolic steroid danazol (400-800 mg/d) has been shown to induce remission in certain patients. Cyclosporine and alfa-interferon have also been used. Plasmapheresis and extracorporeal protein A adsorption have been tried in desperate situations. The autoantibodies responsible for ITP are primarily IgG, and plasmapheresis is of limited value because more than half of the normal IgG pool is in the extravascular space.
Other evolving therapies for refractory ITP include autologous hematopoietic stem cell transplantation and anticytokine therapy with etanercept.
ITP during pregnancy
The optimal management of ITP during pregnancy is considerably controversial. Most pregnant women with ITP are treated with steroids and have relatively few complications involving the fetus and mother. [4]
Patients whose condition is resistant to prednisone can be treated with IVIG. Splenectomy has been performed during pregnancy but should be avoided whenever possible. A platelet count of 50,000/µL is usually sufficient for major surgeries, including splenectomy and cesarean delivery.
The overriding concern is thrombocytopenia developing in the fetus. The IgG autoantibodies in ITP can cross the placenta and may cause thrombocytopenia in the fetus. In most recent studies of ITP complicating pregnancy, severe fetal thrombocytopenia has been uncommon. However, thrombocytopenia is occasionally observed in infants born to mothers who have thrombocytopenia.
No laboratory parameter helps predict the platelet count in the fetus. Previous obstetric history is the only useful predictor. The platelet count of the fetus before delivery can be determined by fetal scalp sampling or percutaneous cord blood sampling.
However, these are invasive procedures associated with serious complications, carrying a risk of intracranial hemorrhage similar to or higher than that of ITP due to neonatal thrombocytopenia. Further, platelets are often clumped, leading to spuriously low platelet count and unnecessary cesarean delivery.
For more information, see Immune Thrombocytopenia and Pregnancy.
Treatment of Thrombotic Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura (TTP) is a medical emergency, and prompt recognition and immediate initiation of plasma exchange is necessary. Until plasma exchange is instituted, fresh frozen plasma should be administered. [42]
Plasma exchange therapy, introduced 30 years ago, has dramatically improved the prognosis for patients with TTP. Current mortality rates remain approximately 20%, compared with the greater than 90% mortality rate observed before the advent of plasma exchange therapy.
Plasma exchange (3-5 L/d) is instituted promptly and continued daily until the patient's platelet count is normalized and the LDH level is within the reference range. Several weeks of plasma exchange may be required before a durable remission is achieved.
Antiplatelet agents have not been shown to alter the natural history of the disease. Milder forms of TTP may respond to steroids.
Rituximab is now routinely recommended for treatment of acute TTP, typically in patients who have a suboptimal response to treatment, but also as first-line therapy. Early administration of rituximab is associated with faster remissions and fewer plasma exchanges. Rituximab therapy may also prevent relapses. [43]
In more severe cases, salvage strategies may include twice-daily therapeutic plasma exchange, pulses of cyclophosphamide, and vincristine. [43] Anecdotal reports indicate cases of TTP responding to a variety of therapies, including the following:
-
IVIG
-
Recombinant ADAMTS13 [43]
-
Inhibitors of the glycoprotein-Ib/IX–von Willebrand factor (vWF) axis [43]
-
Staphylococcal protein A adsorption
-
N-acetylcysteine [44]
-
Intravenous bortezomib [45]
Caplacizumab (Caplivi) is approved for acquired thrombotic thrombocytopenic purpura (aTTP) in combination with plasma exchange and immunosuppressive therapy. Caplacizumab is a monoclonal antibody that targets the A1-domain of vWF, inhibits the interaction between vWF and platelets, and thereby reduces both vWF-mediated platelet adhesion and platelet consumption.
Patients who relapse frequently or patients who require large volumes of replacement therapy are candidates for splenectomy. Splenectomy decreases the rate of relapse in patients with chronic relapsing forms of the disease.
Treatment of von Willebrand Disease
Desmopressin (DDAVP) is a vasopressin analogue that releases vWf from endothelial cells.
Most patients with type I von Willebrand disease can be treated with DDAVP for minor surgeries and dental procedures. The usual dose is 0.3 μg/kg infused slowly approximately 30 minutes before an operative procedure. This dose can be repeated once a day for 2-3 days, after which it is ineffective because of tachyphylaxis. Other adverse effects occasionally include a hypertensive response and hyponatremia.
An intranasal preparation of DDAVP has been made available for individuals with von Willebrand disease and is administered at a dose of 150 μg or 300 μg (ie, 150 μg per nostril). The more diluted preparation is used in patients with diabetes insipidus and does not increase vWf levels.
DDAVP does not usually increase factor VIII levels in patients with type IIA and can induce thrombocytopenia in patients with type IIB or pseudo von Willebrand disease.
Replacement therapy is used for more extensive surgeries or trauma and for patients with type II and type III disease. The treatment of choice is vWf concentrates. Purified plasma-derived concentrates of vWF/FVIII (Humate-P or Alphanate) are heat-treated, and the solvent is extracted; therefore, they are considered safe from viral contamination.
Recombinant von Willebrand factor (Vonvendi) was approved by the US Food and Drug Administration (FDA) in 2015. [46] Approval was expanded in 2018 for perioperative management of bleeding, and in 2022 for routine prophylaxis in patients with severe type 3 vWD who had been receiving on-demand therapy. [47]
A 12-month study of rVWF prophylaxis in 23 patients with severe vWD, 18 of them with type 3, found that patients who had previously received on-demand treatment with vWF products had a lower frequency of spontaneous bleeds requiring vWF treatment, and patients who switched from prophylaxis with plasma-derived vWF experienced similar levels of spontaneous bleeding events requiring treatment. Patients with type 3 vWD did experience some spontaneous bleeding events, but most were mucosal bleeds and/or menorrhagia; no fatal or life-threatening bleeds or spontaneous gastrointestinal bleeds occurred. [48]
The dose of vWf concentrate is calculated based on ristocetin cofactor units (usual dose is 50-100 U/kg). The factor VIII level often rises following the infusion of von Willebrand protein concentrate, and it remains elevated for at least for 40 hours, reflecting the half-life of von Willebrand protein rather than that of factor VIII. The need for further doses is often assessed based on clinical criteria rather than blood test results.
Cryoprecipitate has approximately 100 U of factor VIII per bag and has all multimeric forms of vWf. Despite screening tests, patients have a small risk developing viral infections.
Off-label use of emicizumab, a bispecific, factor VIII-mimetic antibody approved for prophylaxis of hemophilia A, has been reported for treatment of active/recurrent bleeds in patients with type 3 vWD. Published case reports and case series suggest that emicizumab can provide effective hemostatic therapy in this setting. [49]
Highly purified preparations of factor VIII or recombinant factor VIII should not be administered to patients with von Willebrand disease, because these preparations have very little von Willebrand factor.
Treatment of Hemolytic-Uremic Syndrome
Therapy for HUS is directed toward the underlying process. Acute kidney injury and chronic kidney disease are managed with fluid and electrolytes and, if necessary, dialysis. Plasma exchange therapy is often administered, but whether this is beneficial remains unclear. Antiplatelet and anticoagulant therapies have been attempted but do not show a marked benefit.
A role for the complement system in the pathogenesis of HUS is elucidated. Eculizumab, a monoclonal antibody that inhibits the formation of a cell membrane attack complex, is approved for the treatment of all pediatric and adult patients with atypical HUS.
Treatment of Uremic Bleeding
The most effective therapy for this platelet dysfunction is vigorous dialysis. For a more immediate correction of uremic bleeding, DDAVP and cryoprecipitate have also been shown to be useful in providing hemostasis. These modalities provide a short-term benefit until dialysis corrects the hemostatic defects.
In addition to effective dialysis, conjugated estrogen has been shown to decrease bleeding in patients with uremic hemorrhages.
Treatment of Severe Thrombocytopenia With Bleeding
Bleeding in a patient with a very low platelet count is a medical emergency. The presence of hemorrhagic bullae in the buccal mucosa and retinal hemorrhages are harbingers of internal and intracranial bleeding.
Diseases that cause such severe thrombocytopenia are ITP, TTP, posttransfusion purpura, drug-induced thrombocytopenia, and aplastic anemia. Differentiating TTP from ITP is very important, because platelet transfusions are contraindicated in patients with TTP, and plasma exchange therapy should be initiated as soon as possible in patients with TTP.
Careful examination of the peripheral smear helps differentiate ITP from TTP. Furthermore, the presence of neurologic signs, renal failure, fever, and a high LDH level also helps in the diagnosis of TTP.
The patient's medication history should be reviewed, and drug-induced thrombocytopenia should be considered if a temporal relationship exists between the thrombocytopenia and drug exposure.
Patients with liver disease and those who abuse alcohol often present with severe thrombocytopenia following binge drinking. These patients may have severe thrombocytopenia resulting from splenomegaly, alcohol-induced suppression of platelet production, folate deficiency, and DIC from active liver disease.
Aplastic anemia is associated with pancytopenia, and the smear examination findings help differentiate it from ITP.
Once the diagnosis of ITP with clinically significant bleeding is established, treatment with steroids (IV methylprednisolone at 30 mg/kg) and IVIG should be started immediately.
Platelet transfusions are administered to patients with severe clinical bleeding, and a sustained increase in platelet counts is sometimes observed in those with ITP.
Currently, emergency splenectomy is rarely necessary and is only considered before an emergency operation such as evacuation of an intracranial hematoma.
Prognosis
Immune thrombocytopenia
ITP is generally a benign disorder. Severe ITP with a platelet count of less than 5000/µL is occasionally associated with fatal hemorrhages in the brain or internal organs. Risk factors for serious or life-threatening hemorrhage include the following:
-
Advanced age
-
Disease refractory to treatment
-
Previous history of hemorrhage
-
Concomitant bleeding disorders (eg, hemophilia, uremia)
Thrombotic thrombocytopenic purpura
TTP is a very serious disorder. The introduction of plasma exchange therapy has improved the prognosis, but mortality rates remain approximately 20%.
Congenital disorders
Among congenital bleeding disorders involving platelets, type III von Willebrand disease and type I Glanzmann thrombasthenia are severe diseases associated with lifelong hemorrhages. Most other platelet disorders are mild bleeding disorders.
Splenectomized patients
Patients who have undergone splenectomy should be warned about the low risk of severe sepsis following splenectomy. These patients should be immunized with pneumococcal vaccine before splenectomy.
Questions & Answers
Overview
How do platelet disorders occur?
What are major mechanisms of platelet function?
What is the pathophysiology of platelet disorders?
How is immune thrombocytopenia (ITP) characterized?
What are the demographics of acute immune thrombocytopenia (ITP)?
What are the signs and symptoms of acute immune thrombocytopenia (ITP)?
What are the physical exam findings in acute immune thrombocytopenia (ITP)?
What is the most common cause of thrombocytopenia in otherwise healthy children?
What is the relationship between acute leukemia and thrombocytopenia?
What are the demographics of chronic immune thrombocytopenia (ITP)?
What is gestational thrombocytopenia?
What is the role of platelet dysfunction in hypertensive disorders of pregnancy?
What is the pathogenesis of posttransfusion purpura?
What is the role of platelet dysfunction in neonatal alloimmune thrombocytopenia?
How do drugs cause thrombocytopenia?
Which common drugs are associated with thrombocytopenia?
How is drug-induced thrombocytopenia diagnosed?
What are the manifestations of thrombotic thrombocytopenic purpura (TTP)?
What causes thrombotic thrombocytopenic purpura (TTP)?
What is hemolytic-uremic syndrome (HUS)?
What is von Willebrand disease (vWD)?
What is Bernard-Soulier syndrome?
What is Glanzmann thrombasthenia?
How common are functional disorders of platelets?
What is the role of the von Willebrand factor (vWf) in hemostasis?
What are the bleeding manifestations of von Willebrand disease (vWD)?
How does von Willebrand disease (vWD) manifest in pregnancy?
What are the coagulation study abnormalities in von Willebrand disease (vWD)?
What are variants of von Willebrand disease (vWD)?
What causes platelet thromboxane synthesis disorders?
What is the role of enzyme mutations in platelet thromboxane synthesis disorders?
What causes platelet release reaction disorders?
What is the role of platelet dysfunction in uremia?
What is the epidemiology of platelet disorders?
Which features of the history and physical exam are associated with platelet disorders?
What causes platelet disorders?
What are the disorders of platelet function?
Which lab studies are indicated in the workup of platelet disorders?
What are the peripheral smear findings in platelet disorders?
Which lab studies are indicated in the workup of platelet-associated immunoglobulin G?
What is the role of primary hemostasis bleeding time testing in the workup of platelet disorders?
How is platelet aggregation measured in the workup of platelet disorders?
Which imaging studies are indicated in the workup of platelet disorders?
What is the role of bone marrow exam in the workup of platelet disorders?
How effective is eltrombopag for the treatment of chronic immune thrombocytopenia (ITP) in adults?
How effective is romiplostim for the treatment of chronic immune thrombocytopenia (ITP) in adults?
How is chronic immune thrombocytopenia (ITP) in adults treated?
When is splenectomy indicated in the treatment of chronic immune thrombocytopenia (ITP) in adults?
What evolving treatments are available for chronic immune thrombocytopenia (ITP) in adults?
How is immune thrombocytopenia (ITP) managed in pregnancy?
How is thrombotic thrombocytopenic purpura (TTP) treated?
Which salvage strategies are used in the management of thrombotic thrombocytopenic purpura (TTP)?
How is hemolytic-uremic syndrome (HUS) treated?
How is thrombotic thrombocytopenic purpura (TTP) treated?
How is von Willebrand disease (vWd) treated?
How is uremic bleeding in platelet disorders treated?
What is the prognosis of immune thrombocytopenic purpura (ITP)?
What is the prognosis of thrombotic thrombocytopenic purpura (TTP)?
What is the prognosis of congenital platelet disorders?
Does acute immune thrombocytopenia (ITP) in children always require treatment?
What are treatment options for acute immune thrombocytopenia (ITP) in children?
What is the prognosis for children with chronic immune thrombocytopenia (ITP)?
How is alloimmune thrombocytopenia in neonates treated?
-
Normal hemostasis.
-
Purpuric spots.
-
Spurious thrombocytopenia. Peripheral smear from a patient reported to have platelet counts ranging from 10,000 to 150,000/μL on various occasions. The smear shows clumping of the platelets and satellitism involving neutrophils and platelets.
-
Examination of peripheral smears in immune thrombocytopenia often shows giant platelets. These platelets reflect the increased megakaryocytic mass in the marrow.
-
Peripheral smear from a patient with Bernard-Soulier syndrome showing giant platelets. These platelets are not counted as platelets in most particle counters.
-
Peripheral smear in thrombotic thrombocytopenic purpura shows red blood cell fragments and basophilic cells, in addition to thrombocytopenia.
-
Bone marrow in immune thrombocytopenia reveals an increased number of megakaryocytes.
-
Peripheral smear in thrombotic thrombocytopenic purpura shows schistocytes.
-
Normal hemostasis. ADP = adenosine diphosphate; RBC = red blood cell; vWF = von Willebrand factor
-
Platelet activation pathways. 5-HT = 5-hydroxytryptamine; ADP = adenosine diphosphate; ATP = adenosine triphosphate; cAMP = cyclic adenosine monophosphate; CLEC2 = C-type lectin-like receptor 2; DAG = diacylglycerol; GP = glycoprotein; IP3 = inositol trisphosphate; ITAM = immunoreceptor tyrosine-based activation motif; P2Y = purinergic receptor; PAR = protease-activated receptor; PDGF = platelet-derived growth factor; PF4 = platelet factor 4; PIP2 = phosphatidylinositol 4,5-bisphosphate; PKC = protein kinase C; TGF = transforming growth factor; VWF= von Willebrand factor
-
Acquired thrombocytopenias.
-
Platelet aggregation patterns.
Tables

What would you like to print?
- Overview of Platelet Disorders
- Pathophysiology of Platelet Disorders
- Autoimmune Thrombocytopenias
- Disorders of Platelet Function
- Disorders of Secretion and Thromboxane Synthesis
- Platelet Dysfunction in Uremia
- Epidemiology of Platelet Disorders
- Clinical Presentation of Platelet Disorders
- Etiology of Platelet Disorders
- Laboratory Studies
- Imaging Studies
- Bone Marrow Examination
- Treatment of ITP in Children
- Treatment of Chronic ITP in Adults
- Treatment of Thrombotic Thrombocytopenic Purpura
- Treatment of von Willebrand Disease
- Treatment of Hemolytic-Uremic Syndrome
- Treatment of Uremic Bleeding
- Treatment of Severe Thrombocytopenia With Bleeding
- Prognosis
- Questions & Answers
- Show All
- Media Gallery
- References







