Practice Essentials
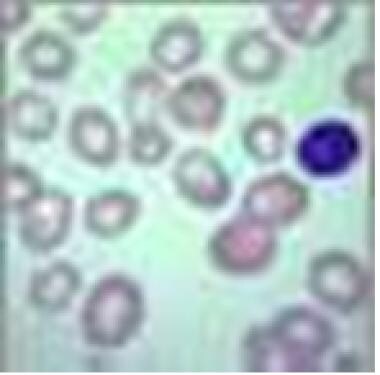
Megaloblasts are large nucleated red blood cells. (See the image below.) Vitamin B12 deficiency (eg, pernicious anemia), folic acid deficiency, and certain medications are the most common causes of megaloblastic anemia, a macrocytic anemia. Although patients may be virtually asymptomatic, since the anemia develops insidiously, those with severe anemia may experience weakness and cardiopulmonary, gastrointestinal, mental, and neurologic signs and symptoms.
See 21 Hidden Clues to Diagnosing Nutritional Deficiencies, a Critical Images slideshow, to help identify clues to conditions associated with malnutrition.
The objectives of this article are to review the pathophysiology, clinical presentation, diagnosis, and management of megaloblastic anemias. An overview of the physiology and biochemistry of vitamin B12 and folate under normal and pathological conditions is included.
Go to Pediatric Megaloblastic Anemia, Anemia, Chronic Anemia, Myelophthisic Anemia, Hemolytic Anemia, and Sideroblastic Anemias for complete information on these topics.
Pathophysiology
Megaloblastosis describes a heterogeneous group of disorders that share common morphologic characteristics: large cells with an arrest in nuclear maturation. Nuclear maturation is immature relative to cytoplasmic maturity. Hence, these cells, which can be seen in bone marrow aspirates and in peripheral smears, have been called megaloblasts. These abnormalities are due to impaired DNA synthesis and, to a lesser extent, RNA and protein synthesis.
Megaloblastic changes are most apparent in rapidly dividing cells such as blood cells and gastrointestinal cells. [1, 2, 3] In addition to large nucleated red blood cells (megaloblasts), hypersegmented neutrophils can be seen on peripheral smears, and giant bands occur in bone marrow.
The common feature in megaloblastosis is a defect in DNA synthesis in rapidly dividing cells. To a lesser extent, RNA and protein synthesis are impaired. Unbalanced cell growth and impaired cell division occur since nuclear maturation is arrested. More mature RBC precursors are destroyed in the bone marrow prior to entering the blood stream (intramedullary hemolysis). [1, 3]
The most common causes of megaloblastosis are vitamin B12 and folate deficiencies, medications, and direct interference of DNA synthesis by HIV infections and myelodysplastic disorders.
Vitamin B12 (cobalamin) and folate biochemistry
Vitamin B12 differs from other water-soluble vitamins in that it is stored in the liver. In addition, vitamin B12 has to be protected during its passage through the gastrointestinal tract to the distal ileum, the site of B12 absorption. The primary sources of cobalamin (Clb), a cobalt-containing vitamin, are meat, fish, and dairy products and not vegetables and fruit. Cyano - Clb, the form used in supplements, is not a natural form but is an in vitro artifact. However, cyano-Clbis readily converted into biologically active forms in humans and other mammals. 5’-Deoxyladenosyl-Clb, methyl-Clb, are the active forms of cobalamin.
Clb is a cofactor for only 2 enzymes in mammals, methionine synthase and L-methylmalonyl-CoA mutase. Methyl-Clb is the cofactor for methionine synthase, and 5’-deoxyladenosyl-Clb is the cofactor for L-methylmalonyl-CoA mutase.
Methionine synthase that requires cofactor methyl-Clb is important for one carbon transfer and is a key enzyme in the methionine cycle. This enzyme is needed to convert homocysteine to methionine involving the transfer of a methyl group. Tetrahydrofolate is a cofactor in this reaction. Methionine, in turn, is required for the synthesis of S-adenosylmethionine (SAM), a methyl group donor used in many biological methylation reactions, including the methylation of sites in DNA and RNA. Diminished activity of methionine synthase or decreased tetrahydrofolate can cause defective DNA maturation and megaloblastic changes. Diminished methionine synthase leads to the “folate trap” in which 5-methyl-THF accumulates and cannot serve as a methyl donor and cannot be converted to the THF needed for methionine synthesis (ie, biological dead end).
L-methylmalonyl-CoA mutase requires cofactor 5-deoxyadenosylcobalamin and catalyzes the conversion of L-methylmalonyl-CoA to succinyl-CoA, a key component of the tricarboxylic acid cycle. This biochemical reaction is important for the production of energy from fats and proteins. Succinyl CoA is also required for the synthesis of hemoglobin, the oxygen carrying pigment in red blood cells. The substrate of methylmalonyl-CoA mutase, methylmalonyl-CoA, is derived from propionyl-CoA from the catabolism of valine, threonine, methionine, thymine, cholesterol, and odd-chain fatty acids.
The mechanisms for patchy demyelination and other neurological consequences of cobalamin deficiency are not well-understood. They appear to be independent and different from those responsible for the development of megaloblastic morphology and anemia. Several theories have been developed for the genesis of cobalamin neuropathy, such as the following [4] :
-
Reduced SAM and resultant abnormal methylation may be responsible. Methylation reactions are needed for myelin maintenance and synthesis.
-
Elevated methylmalonic acid (MMA) may be responsible. Cobalamin deficiency leads to reduced cofactor 5-deoxyadenosylcobalamin that is instrumental in an increase in MMA. Increased MMA is associated with the production of abnormal odd chain and branched chain fatty acids with subsequent abnormal myelination
-
Cobalamin deficiency impacts a network of cytokines and growth factors that can be neurotrophic and others neurotoxic. These factors might play a role in cobalamin related neuropathy. [5]
The sources of folates are ubiquitous, and folate is found in vegetables, fruits, and animal protein. Dietary folic is usually conjugated, polyglutamate folates, and are converted to dihydrofolic acid so they can be absorbed. Dihydrofolate is processed to tetrahydrofolate that participates along with methyl-Clb in the synthesis of methionine. Tetrahydrofolate is conjugated to glutamate to function intracellularly.
Cobalamin transport and uptake
The uptake of cobalamin is complex. Dietary cobalamin binds nonspecifically to dietary proteins. Cobalamin is released from food during gastric digestion at a low pH. The released cobalamin then binds to and is protected by R-proteins. R-proteins have a high affinity for binding cobalamin at a low pH. As cobalamin–R-protein complexes enter the duodenum, cobalamin is released from R-proteins because of the alkaline environment (R-proteins have a low affinity at an alkaline pH) and the presence of pancreatic enzymes. Cobalamin released from R-proteins is free to bind to intrinsic factor (IF), which has a high affinity for binding cobalamin at an alkaline pH.
IF is produced in the gastric fundus and cardia. The role of IF is to stabilize cobalamin and transport it to the terminal ileum. Cobalamin-IF complexes are processed by a receptor, cubulin, in the terminal ileum, and cobalamin is released and absorbed.
The absorbed cobalamin is bound to transcobalamin II (TC II). TC II transports cobalamin to cells that internalize and use cobalamin for DNA synthesis. Transcobalamin I (TC I) might be involved in cobalamin storage and is elevated in leukocytes in patients with chronic myelogenous leukemia.
Storage of cobalamin and folate
Cobalamin is the only water-soluble vitamin stored in the body. About 3 mg of cobalamin are stored, of which 1 mg is stored in the liver. Hence, it takes 3-5 years to develop a vitamin B12 deficiency after a total gastrectomy. In contrast, significant amounts of folate are not stored. Clinical evidence of folate deficiency can occur within a month after folate intake is stopped.
Enterohepatic cycle for cobalamin
Several micrograms of cobalamin are secreted daily in bile and then reabsorbed in the terminal ileum. This enterohepatic cycle can stabilize the daily availability of cobalamin when dietary intake is low.
Uptake of folates
Physiologic folate absorption and transport is receptor mediated. There is no equivalent of IF to stabilize and transport ingested folate. Uptake occurs in the jejunum and throughout the small intestine.
Etiology
Major causes for cobalamin deficiency
Cobalamin deficiency may result from the following:
-
Atrophy or loss of gastric mucosa (eg, pernicious anemia, gastrectomy, ingestion of caustic material, hypochlorhydria, histamine 2 [H2] blockers)
-
Functionally abnormal intrinsic factor (IF)
-
Inadequate dietary intake (ie, vegetarian diet)
-
Inadequate proteolysis of dietary cobalamin
-
Insufficient pancreatic protease (eg, chronic pancreatitis, Zollinger-Ellison syndrome [ZES])
-
Bacterial overgrowth in intestine (eg, blind loop, diverticula) - Bacteria compete with the body for cobalamin
-
Diphyllobothrium latum (fish tapeworm) - Competes with the body for cobalamin
-
Disorders of ileal mucosa (eg, resection, ileitis, sprue, lymphoma, amyloidosis, absent IF-cobalamin receptor, Imerslünd-Grasbeck syndrome, ZES, transcobalamin II [TCII] deficiency, use of certain drugs)
-
Disorders of plasma transport of cobalamin (eg, TCII deficiency, R-binder deficiency)
-
Dysfunctional uptake and use of cobalamin by cells (eg, defects in cellular deoxyadenosylcobalamin [AdoCbl] and methylcobalamin [MeCbl] synthesis)
Pernicious anemia
Pernicious anemia, the best-known cause for cobalamin deficiency, results from autoimmune destruction of gastric parietal cells and subsequent reduction in intrinsic factor (IF) production. Significant amounts of cobalamin are not absorbed in the absence of IF. The term “pernicious anemia” is an anachronism—it dates from the era when treatment had not yet been discovered, and the disease was fatal—but it remains in use.
An increased incidence of pernicious anemia in families suggests a hereditary component to the disease. Patients with pernicious anemia have an increased incidence of autoimmune disorders and thyroid disease, suggesting that the disease has an immunologic component. For example, pernicious anemia may occur together with autoimmune thyroid disease, type 1A diabetes mellitus, alopecia, vitiligo, and chronic atrophic gastritis in type III polyglandular autoimmune (PGA) syndrome—one of a rare group of disorders also known as autoimmune polyendocrine syndromes (APS) and polyglandular failure syndromes. Type III PGA occurs in adults. [6]
Children who develop cobalamin deficiency usually have a hereditary disorder, and the etiology of their cobalamin deficiency is different from the etiology observed in classic pernicious anemia. Congenital pernicious anemia is a hereditary disorder in which an absence of IF occurs without gastric atrophy.
Dietary cobalamin deficiency rarely causes megaloblastic anemia, except in strict vegetarians who avoid meat, eggs, and dairy products. In addition, elderly persons may develop megaloblastic anemia as a result of atrophic gastritis and achlorhydria, which impair the release of cobalamins bound to food and, hence, the availability of cobalamin. [7]
Recommended dietary allowances for vitamin B12 are as follows [8] :
-
Birth to 6 months: 0.4 mcg
-
7–12 months: 0.5 mcg
-
1–3 years: 0.9 mcg
-
4–8 years: 1.2 mcg
-
9–13 years: 1.8 mcg
-
14+ years: 2.4 mcg
-
Pregnancy: 2.6 mcg
-
Lactation: 2.8 mcg
In pancreatic insufficiency, the alkaline environment in the small intestine is insufficient for release of cobalamin from R-proteins and binding to intrinsic factor. In the Zollinger-Ellison syndrome, the acid environment also prevents binding of cobalamin to intrinsic factor. In both conditions, the diminished binding to intrinsic factor interferes with cobalamin absorption.
Disorders of the terminal ileum can result in cobalamin deficiency, because the terminal ileum is the site of uptake of cobalamin-IF complexes. Such disorders include tropical sprue, inflammatory bowel disease, lymphoma, and ileal resection. Tropical sprue is more severe than nontropical sprue (celiac disease) and can be associated with both cobalamin and folate deficiencies. It takes several years for cobalamin deficiency to develop after the onset of these disorders because of the time required to deplete cobalamin reserves.
In the Imerslund-Grasbeck syndrome, there is autoimmune destruction of the ileal receptor, cubulin, for the uptake of cobalamin bound to intrinsic factor.
Blind loop syndrome can result in cobalamin deficiency. Bacterial colonization can occur in intestines deformed by strictures, surgical blind loops, scleroderma, inflammatory bowel disease, or amyloidosis. Bacteria then compete with the host for cobalamin.
The fish tapeworm Diphyllobothrium latum can compete with the host for ingested cobalamin. Diphyllobothriasis most often occurs in people in northern latitudes who eat raw or pickled fish.
Nitrous oxide exposure can cause megaloblastosis by oxidative inactivation of cobalamin. Prolonged exposure to nitrous oxide can lead to severe mental and neurological disorders.
The details of hereditary disorders are beyond the scope of this review, but information can be found in other references. [1, 3]
A partial list of medications that can cause cobalamin deficiency includes the following [1, 9, 10] :
-
Purine analogs (6-mercaptopurine, 6-thioguanine, acyclovir)
-
Pyrimidine analogues (5-fluorouracil, 5-azacytidine, zidovudine)
-
Ribonucleotide reductase inhibitors (hydroxyurea, cytarabine arabinoside)
-
Drugs that affect cobalamin metabolism ( p-aminosalicylic acid, phenformin, metformin)
Major causes for folate deficiency
The daily requirement for adults is about 0.4 mg/d. Storage is limited, and folate deficiency develops about 3-4 weeks after the cessation of folate intake.
Folate content in foods and the preparation of foods are major causes for folate deficiency, especially in elderly persons. Folates are very thermolabile. Therefore, excessive heating can lead to inactivation, especially when foods are excessively diluted in water. In the United States, most people obtain sufficient folate from fortified foods. However, alternative diets may contain little folate.
Increased demand can result in deficiency. There is an increased need for folate in hemolysis, pregnancy, lactation, rapid growth, hyperalimentation, renal dialysis, psoriasis, and exfoliative dermatitis.
Intestinal disorders that impede folate absorption include tropical sprue, nontropical sprue (celiac disease or gluten sensitivity), amyloidosis, and inflammatory bowel disease.
With alcoholism, the bioavailability of folate and folate-dependent biochemical reactions can be impaired.
A partial list of medications that can cause folate deficiency includes phenytoin, metformin, phenobarbital, dihydrofolate reductase inhibitors (trimethoprim, pyrimethamine), methotrexate and other antifolates, sulfonamides (competitive inhibitors of 4-aminobenzoic acid), and valproic acid.
The details of hereditary disorders that cause folate deficiency are beyond the scope of this review, but information can be found in other references). [1, 3, 11, 12]
Other causes for megaloblastosis
Megaloblastosis in HIV infection and myelodysplastic disorders is due to a direct effect on DNA synthesis in hematopoietic and other cells.
Epidemiology
United States statistics
Faulty preparation of foods and folate deficiency during pregnancy are the most common causes of megaloblastic anemias. Pernicious anemia is less common. About 1 in 7,500 people in the United States develops pernicious anemia each year. However, current folate administration during pregnancy and vitamin supplementation in elderly persons has decreased the incidence of megaloblastosis.
International statistics
The frequency of megaloblastosis is highest in countries in which malnutrition is rampant and routine vitamin supplementation for elderly individuals and pregnant women is not available.
Demographics
Pernicious anemia and folate deficiencies usually occur in individuals older than 40 years, and the prevalence increases in older populations. Pernicious anemia is diagnosed in about 1% of people older than 60 years. The incidence is slightly higher in women than in men.
The incidence of pernicious anemia is reported to be higher in Sweden, Denmark, and the United Kingdom than in other developed countries.
Prognosis
The prognosis is favorable if the etiology of megaloblastosis has been identified and appropriate treatment has been instituted. However, patients are at risk for hypokalemia and anemia-related cardiac complications during therapy for cobalamin deficiency.
Folate deficiency during pregnancy can lead to neural tube defects and other developmental disorders in the fetus. However, folate in prenatal vitamins given during pregnancy has reduced these morbidities. [13, 14]
-
Megaloblastic anemia. View of red blood cells
-
Megaloblastic anemia. The structure of cyanocobalamin is depicted. The cyanide (Cn) is in green. Other forms of cobalamin (Cbl) include hydroxocobalamin (OHCbl), methylcobalamin (MeCbl), and deoxyadenosylcobalamin (AdoCbl). In these forms, the beta-group is substituted for Cn. The corrin ring with a central cobalt atom is shown in red and the benzimidazole unit in blue. The corrin ring has 4 pyrroles, which bind to the cobalt atom. The fifth substituent is a derivative of dimethylbenzimidazole. The sixth substituent can be Cn, CC3, hydroxycorticosteroid (OH), or deoxyadenosyl. The cobalt atom can be in a +1, +2, or +3 oxidation state. In hydroxocobalamin, it is in the +3 state. The cobalt atom is reduced in a nicotinamide adenine dinucleotide (NADH)–dependent reaction to yield the active coenzyme. It catalyzes 2 types of reactions, which involve either rearrangements (conversion of l methylmalonyl coenzyme A [CoA] to succinyl CoA) or methylation (synthesis of methionine).
-
Megaloblastic anemia. Inherited disorders of cobalamin (Cbl) metabolism are depicted. The numbers and letters correspond to the sites at which abnormalities have been identified, as follows: (1) absence of intrinsic factor (IF); (2) abnormal Cbl intestinal adsorption; and (3) abnormal transcobalamin II (TC II), (a) mitochondrial Cbl reduction (Cbl A), (b) cobalamin adenosyl transferase (Cbl B), (c and d) cytosolic Cbl metabolism (Cbl C and D), (e and g) methyl transferase Cbl utilization (Cbl E and G), and (f) lysosomal Cbl efflux (Cbl F).
-
Megaloblastic anemia. Cobalamin (Cbl) is freed from meat in the acidic milieu of the stomach where it binds R factors in competition with intrinsic factor (IF). Cbl is freed from R factors in the duodenum by proteolytic digestion of the R factors by pancreatic enzymes. The IF-Cbl complex transits to the ileum where it is bound to ileal receptors. The IF-Cbl enters the ileal absorptive cell, and the Cbl is released and enters the plasma. In the plasma, the Cbl is bound to transcobalamin II (TC II), which delivers the complex to nonintestinal cells. In these cells, Cbl is freed from the transport protein.
-
Peripheral smear of blood from a patient with pernicious anemia. Macrocytes are observed, and some of the red blood cells show ovalocytosis. A 6-lobed polymorphonuclear leukocyte is present.
-
Bone marrow aspirate from a patient with untreated pernicious anemia. Megaloblastic maturation of erythroid precursors is shown. Two megaloblasts occupy the center of the slide with a megaloblastic normoblast above.
-
Response to therapy with cobalamin (Cbl) in a previously untreated patient with pernicious anemia. A reticulocytosis occurs within 5 days after an injection of 1000 mcg of Cbl and lasts for about 2 weeks. The hemoglobin (Hgb) concentration increases at a slower rate because many of the reticulocytes are abnormal and do not survive as mature erythrocytes. After 1 or 2 weeks, the Hgb concentration increases about 1 g/dL per week.







