Practice Essentials
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoma, representing approximately 30% of non-Hodgkin lymphomas (NHLs), [1] and it is rapidly fatal if untreated. See the image below. Most cases respond to standard immunochemotherapy. For nonresponders, who have traditionally had a poor prognosis, alternative therapeutic approaches are increasingly available.
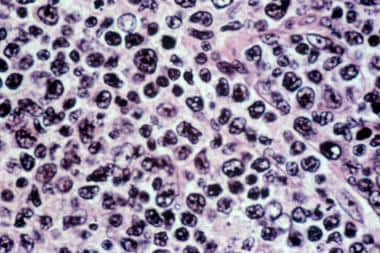 Diffuse large B-cell lymphoma. Hematoxylin and eosin stain of a lymph node biopsy sample showing a mixture of large and small cells. The architecture of the node is lost, with a diffuse pattern of involvement.
Diffuse large B-cell lymphoma. Hematoxylin and eosin stain of a lymph node biopsy sample showing a mixture of large and small cells. The architecture of the node is lost, with a diffuse pattern of involvement.
Signs and symptoms
DLBCLs have a rapid growth rate and present as masses infiltrating tissues or obstructing organs. Signs and symptoms include the following:
-
Pain in an enlarged lymph node or organ: May be noted if the lymphomatous mass enlarges rapidly
-
B symptoms (Ann Arbor staging): Including fever, drenching night sweats, and weight loss
-
Generalized pruritus
-
Anorexia
-
Pedal edema: Caused by extensive pelvic lymphadenopathy
-
Fatigue
-
Chest discomfort or shortness of breath: Caused by mediastinal lymphadenopathy
The following are common findings on physical examination:
-
Lymphadenopathy (ie, cervical, axillary, and inguinal)
-
Splenomegaly
-
Low-grade fever
-
Pedal edema: Resulting from extensive pelvic lymphadenopathy
See Presentation for more detail.
Diagnosis
Lab studies
Lab studies used in the diagnosis and assessment of diffuse large cell lymphoma include the following:
-
Complete blood count: To evaluate involvement of the bone marrow, which may result in anemia, thrombocytopenia, and/or leukopenia
-
Serum electrolyte levels: Electrolyte abnormalities may occur from renal involvement with lymphoma
-
Lactate dehydrogenase and uric acid levels: Elevated levels correspond with the tumor burden
-
Hepatitis B testing: Performed in patients undergoing combination chemoimmunotherapy with rituximab (risk of activation)
-
Flow cytometry: Helps in determining a clonal cell population and in differentiating between B- and T-cell origins
Imaging studies
Imaging studies used in the diagnosis and assessment of diffuse large cell lymphoma include the following:
-
Gastrointestinal imaging: Upper and lower gastrointestinal series indicated in patients with gastrointestinal symptoms, but these studies [2]
-
Central nervous system imaging: Patients with CNS symptoms require brain evaluation with CT scanning with contrast or MRI with gadolinium
-
Bone imaging: Bone scan for patients with unexplained bone pain or elevated alkaline phosphatase levels
-
CT scanning of the neck, chest, abdomen, and pelvis: To help identify degree of lymphadenopathy, presence of extranodal disease, or visceral involvement
-
Gallium-67 scanning - Valuable in staging diffuse large cell lymphomas
-
Multigated acquisition scanning: To evaluate the patient's ejection fraction before chemotherapy
-
Positron emission tomography: To stage disease using fluorodeoxyglucose
Biopsy and lumbar puncture
Bone marrow aspiration and biopsy are performed as part of the staging process to help rule out involvement with lymphoma. Lymph node biopsy is required to establish a definitive diagnosis of NHL. The diagnosis of diffuse large cell lymphoma is usually confirmed after positive findings are obtained from a lymph node biopsy specimen.
In patients with advanced-stage disease, a lumbar puncture for cytologic and chemical analysis of the CSF may be necessary.
See Workup for more detail.
Management
R-CHOP (rituximab plus cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], and prednisone) is the standard chemotherapy regimen for DLBCL; R-CHOP may be followed by radiation therapy (RT). Second-line chemotherapy regimens vary, depending on whether hematopoietic stem cell transplantation (HSCT) is being considered.
Options for relapsed or refractory disease include the following:
-
High-dose chemotherapy with autologous stem cell rescue, with or without RT
-
Alternative chemotherapy regimens
-
Chemoimmunotherapy (eg, antibody-drug conjugates, IgG1-bispecific antibodies)
-
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapy
See Treatment and Medication for more detail.
Background
Historical classification of NHLs
Considerable progress has been made in NHL classification.In 1982, the National Cancer Institute introduced the International Working Formulation, a translation system for other, older classifications, including the Rappaport and the immunologically oriented Lukes-Collins and Kiel systems. The International Working Formulation provided a conceptual framework that groups lymphomas as low grade (indolent), intermediate grade, or high grade, with respect to their natural histories. [3] Of intermediate-grade diffuse large cell lymphomas, approximately 79% were of B-cell origin; 16%, of T-cell origin; and 5%, unclassifiable. Exceptional cases expressed both B-cell and T-cell markers.
In 1994, the International Lymphoma Study Group proposed the Revised European-American Lymphoma (REAL) classification schema. [4] It classifies NHLs as being derived from B or T/natural killer (NK) cells, and it includes disease entities that were not part of the orking formulation.
In addition to morphologic descriptions, the REAL schema included immunologic, cytogenetic, and molecular information to define distinct lymphoma entities. The REAL classification combined the large cell and the immunoblastic categories of diffuse large cell lymphoma. DLBCL is designated under the REAL classification as classic diffuse large cell lymphoma of B-cell origin. Lymphomas of T-cell or NK-cell origin exhibit biologic and clinical features distinct from DLBCLs. (See Workup.)
Currently, the World Health Organization schema is used to classify DLBCLs (see Overview/Pathophysiology).
Pathophysiology
B-cell lymphomas arise at various stages of B-cell development. Under normal circumstances, a pro-B cell undergoes various stages of maturation that include the following:
- The recombination of the V, D, and J gene segments necessary for assembling of the immunoglobulins’ heavy and light chains
- Somatic hypermutation
- Immunoglobulin-class switching
During the process of V(D)J recombination (regulated by the recombination activating genes 1 [RAG1] and 2 [RAG2] enzymes) and somatic hypermutation/immunoglobulin-class switching (regulated by the activation-induced cytidine deaminase [AID] enzyme) phases, multiple DNA alterations occur and normal B cells are susceptible to the development of undesirable chromosomal translocations or gene mutations, leading to the selective growth advantage of a malignant clone and the development of B-cell lymphoma. [5]
The type of mutation(s) and the stage of lymphoid maturation at the time of genetic aberration(s) play a role in the type of lymphoma that may develop in a given patient. [6] Subtypes of diffuse large B-cell lymphoma (DLBCL) arise from genetic alterations occurring during the process of B-cell differentiation/maturation and, in general, are characterized by a blockage of the programmed cell death process (ie, up-regulation of Bcl-2, loss of Bcl-6 function, p53 deletion/mutation), an increase in cell proliferation (eg. increase in nuclear factor kappa B [NFkB], up-regulation of c-Myc), or impaired terminal differentiation (ie, defective Blimp-1 function). Specific genetic alteration(s) or protein expression/function deregulation varies depending on the subtype of DLBCL.
Several oncogenic pathways have been identified in DLBCL (B-cell receptor [BCR] signaling pathway, constitutive activation of NFkB activity pathways, and deregulation of the Bcl-6/apoptosis pathway); however, only one pathway appears to play a pivotal role in the biology of distinct types of DLBCL (ie, germinal center B-cell [GCB] versus activated B-cell [ABC] DLBCL). [5]
On DNA microarray studies, most DLBCLs exhibit gene expression patterns indicative of either of two different stages of B-cell differentiation, which allows classification by cell of origin. [7] GCB DLBCL expresses genes characteristic of germinal center B-cells, while ABC DLBCL expresses genes normally induced during in vitro activation of peripheral blood B-cells. [8] A third heterogeneous subtype of DLBCL does not express genes characteristic of either ABC- or GCB-type cells and is labeled unclassifiable.
GCB DLBCL is associated with recurrent gene translocations involving BCL-2 and C-REL amplification, whereas the ABC subtype has frequent amplifications of the oncogene SPIB, recurrent trisomy for chromosome 3, and activation of the antiapoptotic nuclear factor (NF)-κB signaling pathway. [7]
Constitutive NF kappa-B signaling
Lymphoid malignancies usually avoid cell death by constitutive activation of the NFkB pathway. It is a transcription factor regulating the expression of the immunoglobulin kappa light chain. [9] In B cells, NFkB activation occurs transiently downstream of numerous receptors, including the BCR, CD40, the B-cell–activating factor (BAFF) receptor, and various Toll-like receptors (TLRs). [10] Alternatively, activation of NFkB results from the proteasome degradation of its inhibitor (inhibitor of kappa B [IkB]).
The hallmark of ABC-DLBCL is the activation of NFkB through the classic pathway. Many of the NFkB target genes are expressed in ABC-DLBCL compared with GCB-DLBCL, and this explains how genetic inhibition of this pathway is lethal to ABC- but not GCB-DLBCL lines. [11] Clinically, it has been observed that ABC-DLBCL patients are more refractory to standard immunochemotherapy than other DLBCL subtypes. This could be explained by the ability of NFkB to antagonize the antitumor activity of chemotherapy agents. [12] Moreover, pharmacological inhibitors of NFkB activity (ie, lenalidomide or bortezomib) appear to have selective activity in non–GCB-DLBCL. [13, 14]
Additional mechanisms leading to an increase in NFkB activity have been described in ABC-DLBCL, particularly caspase recruitment domain 11 (CARD11) mutations. The survival of most ABC-DLBCL cell lines depends on the CBM complex (a signaling hub consisting of CARD11, BCL-10, and MALT1). [15] The CBM complex is required for activation of the classic NFkB pathway downstream of the antigen receptors in B and T cells. [16] CARD11 is a multidomain signaling adapter that contains (1) an amino-terminal CARD and coiled-coil domains, (2) an intervening linker domain, and (3) a C-terminal membrane-associated guanylate kinase (MAGUK) domain.
In normal resting conditions, CARD11 is located in the cytosol, where it is presumably kept in an inactive conformation through an intramolecular interaction between its coiled-coil and linker domains. Following signaling via the BCR, protein kinase C (PKC) beta–dependent serine phosphorylation within the CARD11 linker domain occurs and activates CRD11. [17] CARD11 is then able to translocate into the plasma membrane, where it binds to BCL10 and MALT1, forming the CBM complex. Subsequently, the CBM complex plays a pivotal role in the phosphorylation and proteasome degradation of IkB. CARD11 mutations resulting in constitutive engagement of the CBM complex have been described in 10% of ABC-DLBCL patients. [18]
NF kappa-B activation via tonic BCR signaling
Activation of NFkB has also been described in ABC-DLBCL with wild-type CARD11. In this subtype of DLBCL, BCR signaling appears to play a key regulatory role. BCRs present in the surface of B cells are responsible for downstream proliferation and survival signals. [19] The BCR affects B-cell development, antigen-driven clonal selection, and humoral immunity. Structurally, the BCR consists of antigen-binding IgH and immunoglobulin L (IgL) chains noncovalently coupled to CD79A and CD79B subunits. [20] Upon antigen stimulation, clustering of the BCRs occurs, leading to signal transduction via the CD79A and B subunits. [21] CD79A or B mutations have been described in 20% of patients with ABC-DLBCL and lead to the over-expression of CD79 and over-amplification of BCR signaling. [22]
Cell-of-origin classification
Distinct cytogenetic abnormalities have been described in different DLBCL subtypes. The ABC-DLBCL and GCB-DLBCL subtypes in the cell-of-origin (COO) classification have different genetic mutation landscapes, pathobiology, and responses to treatment. [23]
GCB-DLBCL
The most common translocation is t(14,18), with rearrangements of the BCL2 and IGH chain genes. [10] Because of the increased expression of BCL2, the cells are immortalized. Increased BCL2 expression is associated with a poor prognosis and shorter survival. The second most frequent cytogenetic aberration in the GCB subgroup is translocation leading to rearrangement of the MYC gene. A recurrent change noted in a few patients is deletion of the tumour suppressor gene PTEN.
ABC-DLBCL
The most common aberration in ABC-DLBCL is translocation involving the BCL6 gene. [11] Another frequent aberration in the ABC group is trisomy 3. [12] Approximately 18 % of the patients diagnosed with ABC-DLBCL are diagnosed to have a deletion of the tumor suppressor gene P53. Inactivation of P53 results in uncontrolled cell proliferation and subsequent tumor genome instability. Mutations or deletions of P53 decrease the overall survival of all DLBCL patients.
World Health Organization classification
In addition to the GCB and ABC forms of DLBCL not otherwise specified (NOS), the World Health Organization (WHO) 2016 classification of lymphoid neoplasms lists the following subtypes of DLBCL [1] :
-
T-cell/histiocyte-rich large B-cell lymphoma
-
Primary DLBCL of the central nervous system (CNS)
-
Primary cutaneous DLBCL, leg type
-
Epstein-Barr virus–positive (EBV+) DLBCL, NOS
-
EBV+ mucocutaneous ulcer
-
DLBCL associated with chronic inflammation
-
Lymphomatoid granulomatosis
-
Primary mediastinal (thymic) large B-cell lymphoma
-
Intravascular large B-cell lymphoma
-
ALK-positive B-cell lymphoma
-
Plasmablastic lymphoma
-
Primary effusion lymphoma
-
Human herpesvirus 8 (HHV8)-positive DLBCL
Etiology
B-cell restricted markers (CD19, CD20, CD22) are expressed consistently in diffuse large cell lymphoma. Activation antigens are variably expressed by diffuse large B-cell lymphomas (DLBCLs), with human leukocyte antigen (HLA)-DR being the most frequent and CD23 being expressed uncommonly (0-25%). The presence of CD10 or CD5 suggests that at least one third of diffuse large cell lymphomas may have transformed from follicular lymphomas or a small lymphocytic lymphoma.
The majority of DLBCLs demonstrate rearrangements of the immunoglobulin genes by deoxyribonucleic acid (DNA) hybridization techniques, proving their B-cell lineage.
Mutations or allelic losses of the TP53 tumor suppressor gene or 17p13.1 are common in diffuse large cell lymphomas, particularly in the immunoblastic type. Changes in TP53 appear to be particularly involved in the evolution of follicular lymphoma to diffuse large cell lymphoma. [24] A number of cytogenetic abnormalities have been reported in these neoplasms, including t(14;18), t(8;14), trisomy 12, and deletion of 6q. [25, 26]
A study by Pasqualucci et al found that the DLBCL coding genome contains on average more than 30 clonally represented gene alterations per case. Mutations identified included those regulating chromatin methylation (MLL2, seen in 24% of cases) and immune recognition by T cells. [27]
Alizadeh et al concluded that the measurement of LMO2 and TNFRSF9 can be used to predict overall survival in patients with diffuse large cell lymphomas. [28]
Associated conditions
Non-Hodgkin lymphomas (NHLs) have been associated with the following conditions, drugs, and chemical agents:
-
Hereditary immunodeficiency disorders such as ataxia-telangiectasia syndrome, Bruton-type agammaglobulinemia, severe combined immunodeficiency (SCID), Wiskott-Aldrich syndrome, Duncan syndrome, and Chediak-Higashi syndrome
-
Infections such as with human immunodeficiency virus (HIV), Epstein-Barr virus (EBV), Helicobacter pylori, hepatitis C virus (HCV), human T-cell leukemia virus (HTLV), and human herpes viruses (HHVs) [29]
-
Autoimmune disorders such as rheumatoid arthritis, Sjögren syndrome, and systemic lupu s erythematosus
-
Use of drugs such as immunosuppressants and chemotherapeutic agents [29]
-
Occupational factors - These present a weak or an inconsistent risk
Epidemiology
Occurrence in the United States
After a striking increase in incidence rates between 1970 and 1995 (which may in part have reflected improved diagnosis), the rates of new non-Hodgkin lymphoma (NHL) cases stabilized. From 2010-2019, rates of new cases fell on average 1.0% each year; and from 2011-2020, death rates fell on average 2.2% each year. The current US age-adjusted rate is 17.2 cases per 100,000 person-years for both sexes. [31] The estimated rate for diffuse large B-cell lymphomas is approximately 4.68 cases per 100,000 person-years.
It is estimated that approximately 80,550 new cases of NHL will be diagnosed and 20,180 patients will die from NHL in 2023, despite currently available treatment. [32] Lymphomas are a heterogeneous group of malignancies with diverse biology, clinical behavior, and prognosis.
In general, lymphomas can be divided into two groups, Hodgkin lymphoma (HL) and NHL. While infrequent, HL (8,480 estimated new cases in 2020) is commonly diagnosed in younger patients and is curable with appropriate therapy in 85% of cases. In contrast, NHL is the seventh most common cancer in the United States, accounting for 4.3% of all cancers, and the eighth leading cause of cancer deaths, accounting for 3.3% of cancer-related deaths. [31] Diffuse large B-cell lymphoma (DLBCL) is the most common type of NHL diagnosed in the Western hemisphere, representing 30-40% of all NHL cases diagnosed every year in the United States. [33]
DLBCL typically affects patients in their sixth decade, except for primary mediastinal DLBCL variant, which affects mostly females in their late 20s or early 30s. Over the past decades, the incidence of DLBCL has been increasing, a trend that has been independent of the human immunodeficiency virus (HIV) infection epidemic. [34]
International occurrence
In general, the age-adjusted incidence of diffuse large cell lymphomas is higher in developed countries. For males, it varied from 3.7 to 14 cases per 100,000 persons per year from 1983 to 1987. Since the late 20th century, rates for men and women have increased by 50% or more in 20 different countries. [2]
The rates by subtype, such as the subtypes Burkitt lymphoma (Epstein-Barr virus [EBV]–associated lymphoma) and human T-cell leukemia virus (HTLV) type 1–associated lymphoma/leukemia, also vary widely in different geographic areas, with specific subtypes being much more frequent in their endemic areas.
Race-, sex-, and age-related demographics
White individuals have higher rates than people of African or Asian descent [35] ; the Surveillance, Epidemiology, and End Results (SEER) registry demonstrates rates in white men that are 49% higher than in black men, 54% higher than in Japanese American men, and 27% higher than in Chinese American men. [36] These differences also apply to women.
A study by Flowers et al found differences between white and black patients with regard to presentation by and survival rate for individuals with DLBCL. According to the study (a retrospective cohort analysis of 533 white patients and 144 black patients), the median age of diagnosis was 50 years for black patients and 57 years for white patients. A higher percentage of black patients presented with elevated lactate dehydrogenase (LDH) levels, while more whites had a family history of lymphoma than did black patients (8% vs 3%, respectively). [37]
In the study, the survival rate among black patients was lower than among white patients, but both groups demonstrated an improved survival rate with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, prednisone) therapy.
DLBCL affects females more often than males.
Although DLBCL can occur at any age, they generally develop in middle-aged and older adults. Most patients are diagnosed during the seventh or eighth decade of life, with a median age of 63 years.
Prognosis
For non-Hodgkin lymphoma (NHL) overall, 5-year relative survival is 73.2%, based on 2011-2017 data. [31] Data suggest that 5-year survival rates in diffuse large B-cell lymphoma (DLBCL) are higher for white persons than they are for people of African descent, which may or may not reflect socioeconomic factors. Women also have a better survival outcome, as do patients younger than 65 years. [38]
The clinical outcome of lymphoma patients has improved over the last decades as a result of several factors that include the following:
-
A better understanding of the pathogenesis and biology of lymphoid malignancies
-
Advances in technology resulting in a more precise diagnosis (ie, immunophenotyping, cytogenetic or gene expression profiling studies) and staging (ie, functional imaging)
-
The identification and validation of clinically based score indices or biomarkers capable of predicting clinical outcomes and/or response to therapy
-
The adoption of high-dose chemotherapy and autologous stem cell support (HDC-ASCS) for patients with relapsed/refractory disease
-
The development and adoption of novel and effective agents in the management of lymphoid malignancies (eg, monoclonal antibodies).
Risk stratification and prognostic markers in DLBCL
Risk stratification plays an important role in the management of patients with DLBCL and should be performed before starting therapy.
The International Prognostic Index (IPI) score system was the result of a collaborative effort of 16 institutions in Europe and North America that used a dataset containing clinical information of almost 2000 patients. [39] Briefly, the IPI score system is calculated by the sum of the presence or absence of 5 variables easily available in most clinical practices (age ≥ 65 y, performance status ≥ 2, elevated lactate dehydrogenase (LDH), Ann Arbor stage III or IV, and ≥2 extranodal sites of disease). Based on the total score, DLBCL patients are assigned into 4 risk category groups (low, low-intermediate, high-intermediate, and high) with overall survival ranging from 23-75%.
The IPI score has been validated in multiple clinical trials before and after the incorporation of rituximab into the frontline therapy of patients with DLBCL. The IPI score has also been validated in relapsing aggressive NHL. [40]
In addition, modifications from the original predictive score have been formulated, such as the age-adjusted IPI score for patients younger than 65 years and the rituximab-IPI score with similar prognostic power.
While the clinical value of the IPI score is extremely important, especially when analyzing results across multiple clinical trials, it does not provide insightful information in regard to disease biology, including mechanisms of resistance to active treatments. This fact stresses the need to further identify and validate more biologically representative biomarkers of disease response using novel technology such as gene expression profiling (GEP), proteomics, or comparative chromosomal analysis.
In a large multi-center cohort, Alinari et al reported that patients with de novo CD5+ DLBCL have a poor prognosis despite initial rituximab-containing chemotherapy. Moreover, their results suggested that stem cell transplantation fails to salvage the majority of these patients [41] .
Prognostic factors in early-stage disease
A number of studies have analyzed factors predicting better or worse survival rates for patients with limited-stage diffuse large cell lymphoma (stage IA and IIA, nonbulky) treated with combined modality programs.
In a study by the Southwestern Oncology Group (SWOG), a subgroup analysis showed that the 5-year survival rate was better in patients who had a favorable IPI score. [42] Similar results were found in a study of 308 patients with limited disease treated with 3 cycles of a doxorubicin-containing regimen followed by radiotherapy.
In an Eastern Cooperative Oncology Group (ECOG) study, the 6-year disease-free survival rate for patients who achieved complete remission was greater in patients who received chemotherapy plus radiation therapy than it was in patients who received only chemotherapy (73% vs 56%, respectively). [43] The study compared 8 courses of a regimen of cyclophosphamide, Adriamycin, vincristine, prednisone (CHOP), with or without radiation, in patients with previously untreated bulky or extranodal stage I or II diffuse large cell lymphoma.
Despite the differences in disease-free survival, overall survival rates in the 2 groups were similar (64% for the radiation group vs 60% for the other patients). [43] Patients with 3 or more disease sites or a poor performance status were more likely to have treatment failure with the CHOP regimen, with or without radiotherapy.
The results from all of these studies suggest that combined modality therapy can be used to successfully treat patients with limited stage I disease and a stage-modified IPI score of zero. This approach appears to be less successful in patients with bulky stage I or II disease, 3 or more involved disease sites, and/or a stage-modified IPI score of 1 or more.
Prognostic factors in advanced-stage disease
The International Non-Hodgkin Lymphoma Prognostic Factors Project developed a predictive model of outcome for aggressive NHL; ie, stage II bulky or stage III or IV. [39] The following 5 pretreatment characteristics showed independent statistical significance for higher-risk disease:
-
Age older than 60 years
-
Tumor stage III or IV (advanced)
-
More than 1 extranodal site involved by disease
-
Patient performance status of 2 or more
-
LDH elevation above the reference range
Based on those 5 characteristics, patients were stratified into 4 categories, as follows:
-
Low-risk patients - 0 or 1 adverse factor
-
Low-risk to intermediate-risk patients - 2 factors
-
High-risk to intermediate-risk patients - 3 factors
-
High-risk patients - 4 or 5 factors
On analysis by risk stratification, patient outcomes differed with regard to complete response, disease-free survival, and overall survival. For example, patients with a low risk had a complete response rate of 87% and a 5-year survival rate of 73%, as compared with a complete response rate of 44% and a 5-year survival rate of 26% in the high-risk group.
Subsequent studies have confirmed the reproducibility of the IPI for predicting clinical outcome for patients with DLBCL. Currently, poor-risk patients (despite achieving complete response) may be considered for aggressive therapy with HDC-ASCS in first remission.
The International Prognostic Index (IPI), developed by the International Non-Hodgkin's Lymphoma Prognostic Factor Project, is a model for predicting outcomes in patients with aggressive non-Hodgkin lymphoma on the basis of the patients' clinical characteristics before treatment.
Calculators based on the IPI include the foillowing:
-
Revised International Prognostic Index, developed to predict the outcome of patients receiving rituximab with chemotherapy. Diffuse Large B-Cell Lymphoma Prognosis (R-IPI)
-
Event-free survival at 24 months from diagnosis: Diffuse Large B-cell Lymphoma Prognosis (IPI24)
-
Enhanced IPI score, using the National Comprehensive Cancer Network database of 1935 patients treated in the rituximab era: Diffuse Large B-cell Lymphoma Prognosis (NCCN-IPI)
Patient Education
Patients with diffuse large B-cell lymphoma should receive information about the following:
-
Febrile neutropenia
-
Postchemotherapy thrombocytopenia and the tendency to bleed with minimal trauma
-
Chemotherapy-associated alopecia
-
Avoidance of pregnancy in reproductive-aged women
-
Chemotherapy-induced nausea and vomiting
-
Chemotherapy-associated menstrual dysregulation (females) and the possibility of sexual dysfunction
-
Other sequelae of chemotherapeutic agents, Including secondary leukemias, myelodysplastic syndrome, anaphylactic reactions, and potentially fatal infections
-
Fatigue
-
Sperm banking and risk of sterility for males
Clearly explain transfusions (red blood cells and platelets) and associated complications. In addition, discuss the possibility of (1) long-term complications of higher doses of chemoradiotherapy and (2) mortality rates as high as 3-5% from the conditioning regimen in patients who require HDC and ASCT.
For patient education information, see Leukemia and Lymphoma and Non-Hodgkin Lymphoma.
-
Biopsy of a cervical lymph node showing infiltration with a population of large cells (B cells) consistent with diffuse large cell lymphoma.
-
Computed tomography (CT) scan of the abdomen showing mesenteric and retroperitoneal adenopathy in a patient with diffuse large cell lymphoma.
-
Diffuse large B-cell lymphoma. Hematoxylin and eosin stain of a lymph node biopsy sample showing a mixture of large and small cells. The architecture of the node is lost, with a diffuse pattern of involvement.
-
Patient with diffuse large B-cell lymphoma with extranodal involvement. This computed tomography (CT) scan shows an enlarged spleen and liver as a result of lymphomatous involvement.
-
Patient with diffuse large B-cell lymphoma with extranodal involvement (same patient as in the previous image). This patient has an enlarged spleen and liver as a result of lymphomatous involvement. Extensive retroperitoneal lymphadenopathy is also present.
-
This image depicts gallium scans performed as part of a staging workup for a patient with diffuse large B-cell lymphoma. The scans show extensive hepatosplenic and multiple sites of nodal involvement with a gallium-avid tumor.
-
Role of B-cell receptor signaling in promoting proliferation and survival of DLBCL. BCR is the main factor in B-cell biology, playing a key role in B-cell development, antigen-driven clonal selection, and humoral immunity. B-cell receptor signaling activates PI3K-mediated activation of the kinase AKT, which activates many downstream signaling pathways. All these downstream pathways are essential for the survival of B cells. PIP3 is generated as a result of BCR-dependent PI3K activation. BTK also hydrolyzes PIP2 into DAG and IP3. IP3 induces release of calcium stores from the endoplasmic reticulum. Ca and DAG activate PKC, which leads to activation of NF-k B pathyway. PI3K, phosphatidylinositide-3 kinase; AKT, protein kinase B; PTEN, phosphatase and tensin homolog; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-4,5-trisphosphate; IKK, IkB kinase; mTOR, mammalian target of rapamycin; FoxO, Forkhead box transcription factors; GSK3b, glycogen synthase 3-beta; p21, inhibitor of cyclin-dependent kinases.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Guidelines
- Medication
- Medication Summary
- Antineoplastics, Other
- Corticosteroids
- Monoclonal Antibodies
- Antineoplastics, PI3K Delta Inhibitors
- CAR T-cell Therapy
- Selective Inhibitors of Nuclear Export (SINE)
- Antineoplastics, Anti-CD19 Monoclonal Antibodies
- Antineoplastics, Anti-CD20 Monoclonal Antibodies
- Colony-Stimulating Factors Growth Factor
- Antibiotics
- Vitamins
- Antidotes, Other
- Antifungals, Other
- Antiemetics
- Show All
- Questions & Answers
- Media Gallery
- References






