Practice Essentials
Hemolysis is the premature destruction of erythrocytes. A hemolytic anemia will develop if bone marrow activity cannot compensate for the erythrocyte loss. The clinical severity of the anemia depends on whether the onset of hemolysis is gradual or abrupt as well as the extent of erythrocyte destruction. Mild hemolysis can be asymptomatic while the anemia in severe hemolysis can be life threatening and lead to angina and cardiopulmonary decompensation.
The clinical presentation also reflects the underlying cause for hemolysis. For example, sickle cell anemia (see the image below) is associated with painful vaso-occlusive crises. (See Presentation.)
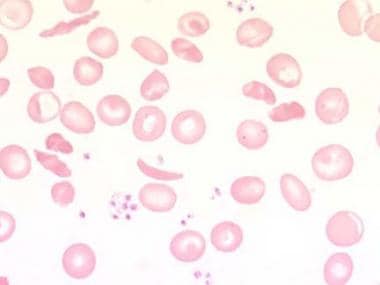 Peripheral blood smear with sickled cells at 1000X magnification. Image courtesy of Ulrich Woermann, MD.
Peripheral blood smear with sickled cells at 1000X magnification. Image courtesy of Ulrich Woermann, MD.
Hemolytic anemia has multiple causes, and the clinical presentation can differ depending on the etiology. An array of laboratory tests are available for detecting hemolysis, and specialized tests may be indicated to diagnose the cause of hemolysis (see Workup). There are differences in the management of various types of hemolytic anemias (see Treatment).
Go to Anemia, Iron Deficiency Anemia, and Chronic Anemia for complete information on these topics.
Pathophysiology
Hemolysis can be due to hereditary and acquired disorders. [1, 2] The etiology of premature erythrocyte destruction is diverse and can be due to conditions such as intrinsic membrane defects, abnormal hemoglobins, erythrocyte enzymatic defects, immune destruction of erythrocytes, mechanical injury, and hypersplenism.
Hemolysis can occur intravascularly or extravascularly. Autoimmune hemolytic anemia and hereditary spherocytosis are examples of extravascular hemolysis because the red blood cells are destroyed in the spleen and other reticuloendothelial tissues. [3] Intravascular hemolysis occurs in hemolytic anemia due to the following:
-
Prosthetic cardiac valves
-
Transfusion of ABO incompatible blood
Hemolysis may also be intramedullary, when fragile red blood cell (RBC) precursors are destroyed in the bone marrow prior to release into the circulation. Intramedullary hemolysis occurs in pernicious anemia and thalassemia major. Skull and other skeletal deformities can occur in childhood due to a marked increase in hematopoiesis and resultant bone marrow expansion in disorders such as thalassemia.
Hemolysis is associated with a release of RBC lactate dehydrogenase (LDH). Hemoglobin released from damaged RBCs leads to an increase in indirect bilirubin and urobilinogen levels.
A patient with mild hemolysis may have normal hemoglobin levels if increased RBC production matches the rate of RBC destruction. However, patients with mild hemolysis may develop marked anemia if their bone marrow erythrocyte production is transiently shut off by viral (parvovirus B19) or other infections. This scenario would be an aplastic crisis since the bone marrow can no longer compensate for ongoing hemolysis.
Etiology
A wide range of causes of hemolytic anemia have been documented. [7, 8, 9, 10, 11, 12, 13, 14, 15, 1, 16] Only the more commonly encountered hemolytic disorders are discussed in this article.
Hereditary disorders may cause hemolysis as a result of erythrocyte membrane abnormalities, enzymatic defects, and hemoglobin abnormalities. Hereditary disorders include the following:
Acquired causes of hemolysis include the following:
-
Immune disorders
-
intravenous immunoglobulin G (IVIG) [17]
-
Antiviral agents (eg, ribavirin [19] )
-
Physical damage
-
Post-valve replacement [22]
-
Contrast medium iomeprol [23]
Autoimmune hemolytic anemia (AIHA) can be due to warm or cold autoantibody types and, rarely, mixed types. [16, 24, 25, 26] Most warm autoantibodies belong to the immunoglobulin IgG class. These antibodies can be detected by a direct Coombs test, which also is known as a direct antiglobulin test (DAT). AIHA may occur after allogeneic hematopoietic stem cell transplantation. The 3-year cumulative incidence in this population has been reported at 4.44%. [27]
AIHA is rare in children and has a range of causes. Autoimmune hemolysis can be primary or secondary to conditions such as infections (viral, bacterial, and atypical), systemic lupus erythematosus (SLE), autoimmune hepatitis (AIH), and H1N1 influenza. H1N1 influenza–associated AIHA in children may respond to treatment with oseltamivir and intravenous immunoglobulin. [9]
Fetal splenomegaly and associated hepatomegaly could be due to hemolysis, but infections are the most likely cause. Congestive heart failure and metabolic disorders should be considered. Rarely, leukemia, lymphoma, and histiocytosis are associated with splenomegaly. [28]
Microangiopathic hemolytic anemia, which results in the production of fragmented erythrocytes (schistocytes), may be caused by any of the following [29, 30] :
-
Defective prosthetic cardiac valves
-
Disseminated intravascular coagulation (DIC)
-
Hemolytic uremic syndrome (HUS)
-
Thrombotic thrombocytopenic purpura (TTP)
In paroxysmal nocturnal hemoglobinuria, hemolysis is due to intravascular complement-mediated destruction of erythrocytes.
Epidemiology
Hemolytic anemia represents approximately 5% of all anemias. Acute AIHA is relatively rare, with an incidence of one to three cases per 100,000 population per year. [31]
A review of the Nationwide Inpatient Sample database found that the prevalence of nonimmune hemolytic anemia was 0.17% in all hospitalized patients with alcoholic liver disease. The presence of anemia among inpatients with alcoholic liver disease was associated with a significantly worse prognosis, including longer average length of stay (8.8 vs. 6.0 d1), increased hospital charges ($38,961 vs. $25,244), and higher mortality (9.0% vs. 5.6%). [32]
Hemolytic anemias are not specific to any race. However, sickle cell disorders are found primarily in Africans, African Americans, some Arabic populations, and Aborigines in southern India.
Several variants of G6PD deficiency exist. The A(-) variant is found in West Africans and African Americans. Approximately 10% of African Americans carry at least 1 copy of the gene for this variant. The Mediterranean variant occurs in individuals of Mediterranean descent and in some Asians.
Most cases of hemolytic anemia are not sex specific. However, AIHA is slightly more likely to occur in females than in males. G6PD deficiency is an X-linked recessive disorder and therefore primarily males are affected while females are more commonly carriers.
Although hemolytic anemia can occur in persons of any age, hereditary disorders are usually evident early in life. AIHA is more likely to occur in middle-aged and older individuals.
Prognosis
The prognosis for patients with hemolytic anemia depends on the underlying cause.
Overall, mortality rates are low in hemolytic anemias. However, the risk is greater in older patients and patients with cardiovascular impairment.
Morbidity depends on the etiology of the hemolysis and the underlying disorder, such as sickle cell anemia or malaria.
Patient Education
Patients should be able to identify symptoms and signs of the recurrence of hemolysis. They should seek prompt medical attention if symptoms reoccur.
Patients with G6PD deficiency should know which medications to avoid.
For patient education information, see Anemia.
-
Polychromasia.
-
Spherocytes. One arrow points to a spherocyte; the other, to a normal RBC with central pallor.
-
Schistocytes (thrombotic thrombocytopenic purpura).
-
Peripheral blood smear with sickled cells at 1000X magnification. Image courtesy of Ulrich Woermann, MD.
-
Supravital stain in hemoglobin H disease reveals Heinz bodies (golf ball appearance).
-
Hemolytic anemia algorithm.







