Practice Essentials
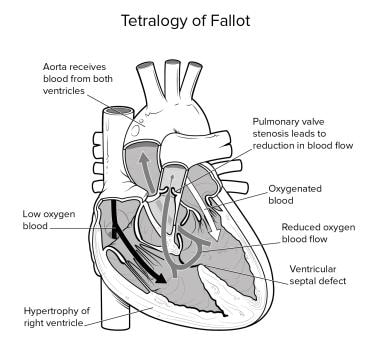
Tetralogy of Fallot (TOF) (pronounced te-tral-uh-jee of Fal-oh), one of the most common congenital heart disorders, comprises right ventricular (RV) outflow tract obstruction (RVOTO) (infundibular stenosis), ventricular septal defect (VSD), aorta dextroposition (overriding aorta), and RV hypertrophy (see the image below). The mortality rate in untreated patients reaches 50% by age 6 years, but in the present era of cardiac surgery, children with simple forms of tetralogy of Fallot enjoy good long-term survival with an excellent quality of life.
However, it is important to understand that corrective surgery for tetralogy of Fallot performed in childhood is not curative surgery. As a result, many children with repaired tetralogy of Fallot survive into adulthood and are seen at cardiology clinics. [1, 2] Some patients who never underwent surgery for mild tetralogy during childhood may present as adults with a variety of symptoms, and other patients who may only have had a palliative procedure (eg, shunt placement) can also present as adults. [3]
Signs and symptoms of tetralogy of Fallot
The clinical features of tetralogy of Fallot in the adult are directly related to the severity of the anatomic defects and may include the following:
-
Lack of exercise endurance
-
Palpitations
-
Gradual decline in bodily functions
Infants, however, often display the following:
-
Difficulty with feeding
-
Failure to thrive
-
Episodes of bluish pale skin during crying or feeding (ie, "Tet" spells)
-
Exertional dyspnea, usually worsening with age
Physical findings in adults include the following:
-
Exertional dyspnea
-
Syncope
-
Palpitations
-
Evidence of right-sided heart failure (eg, elevated jugular venous pressure [JVP], ascites, peripheral edema, hepatomegaly)
-
Large "A wave" in JVP tracings
-
In individuals with pulmonary valve insufficiency: Low-pitched, short diastolic murmur; may have an extra heart sound (ejection click); may have a murmur
-
RV dysfunction, arrhythmias
See Presentation for more detail.
Diagnosis of tetralogy of Fallot
In adult patients, the following laboratory studies may be helpful:
-
Complete blood cell count: A mild anemia may be present; polycythemia is rare in the absence of cyanosis.
-
Coagulation profile: May be abnormal in patients with cyanosis and bleeding
-
Arterial blood gas (including serum lactate, base excess, oxygen partial pressure [PO2]): The patient's oxygenation status, serum lactate levels, and base excess value appear to be prognostic markers for mortality in those undergoing surgical repair of tetralogy of Fallot. [4]
-
Blood cultures: Obtain blood cultures in febrile patients to rule out endocarditis or sepsis.
Imaging studies include the following:
-
Chest radiographs
-
Echocardiography
-
Magnetic resonance imaging (MRI)
Chest radiographs have the following attributes:
-
May be normal or depict cardiomegaly and prominent RV shadowing
-
Although the classic boot-shaped heart (coeur en sabot) is the hallmark of the disorder in infants, this shape of the heart may not be seen in adult patients.
Echocardiography has the following attributes:
-
Color-flow Doppler echocardiography: Assesses overall cardiac function and the status of the valves, and for the presence of any residual VSD, ductus arterosus, or atrial septal defect
-
Allows evaluation of the valvular anatomy but is unable to visualize the coronary anatomy in adults
-
Reveals the grade and severity of any RVOT obstruction
MRI has the following attributes:
-
Gold standard for assessing RV function and size, and for quantifying the pulmonary regurgitant volume
-
Maps the velocity of pulmonary regurgitation and provides good delineation of the aorta size and assessment of the pulmonary arteries and its branches, the status of the RVOT, and the presence of VSDs, and/or RV hypertrophy [5]
-
Can also be used to measure intracardiac pressures, gradients, and blood flows
Cardiac catheterization is useful in any of the following settings:
-
When the anatomy cannot be completely defined by echocardiography
-
When disease in the pulmonary arteries is a concern
-
When pulmonary vascular hypertension is possible
Cardiac catheterization allows the following:
-
Assessment of the pulmonary annulus size and pulmonary arteries
-
Assessment of the severity of RVOTO
-
Location of the position and size of the VSD
-
Ruling out possible coronary artery anomalies
See Workup for more detail. See also the Guidelines section for the 2018 American Heart Association/American College of Cardiology (AHA/ACC) recommendations for the management of adults with tetralogy of Fallot.
Treatment of tetralogy of Fallot
For adults with acute cyanosis, place them in a knee-chest position. In addition, provide/administer the following:
-
Oxygen
-
IV morphine
-
IV propranolol for severe cases [6]
Most symptomatic adults with tetralogy of Fallot require some type of surgical intervention. Pulmonary valve replacement is the most common procedure performed, usually under cardiopulmonary bypass.
Factors that increase the risk for surgery in adult patients with tetralogy of Fallot include the following:
-
Cardiogenic shock
-
Poor RV function
-
Diminutive pulmonary arteries
-
Other major associated anomalies, such as tricuspid atresia or an anomalous coronary artery
-
Multiple previous surgeries
-
Advanced age
-
Severe annular hypoplasia
-
High peak RV–to–left ventricular pressure ratio
-
Multiple ventricular septal defects (VSDs)
-
Right-sided heart failure
See Treatment and Medication for more detail, as well as the Guidelines section for the 2018 American Heart Association/American College of Cardiology (AHA/ACC) recommendations for the management of adults with tetralogy of Fallot.
Background
Tetralogy of Fallot (TOF) (pronounced te-tral-uh-jee of Fal-oh) is one of the most common congenital heart disorders (CHDs). This condition is classified as a cyanotic heart disorder, because tetralogy of Fallot results in an inadequate flow of blood to the lungs for oxygenation (right-to-left shunt) (see the following image). Patients with tetralogy of Fallot initially present with cyanosis shortly after birth, thereby attracting early medical attention. In most cases, the cyanosis does not present for a few weeks or months after birth; however, infants born with transposition of the great vessels usually present with cyanosis immediately after birth.
Typical features
The four features typical of tetralogy of Fallot include right ventricular (RV) outflow tract obstruction (RVOTO) (infundibular stenosis), ventricular septal defect (VSD), aorta dextroposition (overriding aorta), and right ventricular hypertrophy. Occasionally, a few children also have an atrial septal defect (ASD), which makes up the pentad of Fallot. The basic pathology of tetralogy is due to the underdevelopment of the RV infundibulum, which results in an anterior-leftward malalignment of the infundibular septum. This malalignment determines the degree of RVOTO.
The clinical features of tetralogy of Fallot are generally typical, and a preliminary clinical diagnosis can almost always be made. Because most infants with this disorder require surgery, it is fortunate that the availability of cardiopulmonary bypass (CPB), cardioplegia, and surgical techniques is now well established. Most surgical series report excellent clinical results with low morbidity and mortality rates.
Surgery in infancy
The first surgery to repair tetralogy of Fallot consisted of placement of a shunt to relieve the cyanosis. Primary repair is currently recommended within the first 12 months of life; in general, excellent results are obtained at most centers. Ever since primary repair of tetralogy of Fallot became the standard of care nearly 30 years ago, more adult patients with repaired tetralogy of Fallot in early childhood are living longer, [1, 2] and they are being seen at many cardiac clinics. Some patients who never underwent surgery for mild tetralogy during childhood may present as adults with a variety of symptoms, and other patients who may only have had a palliative procedure (eg, shunt placement) can also present as adults. [3]
What happens years after surgical repair?
It is important to understand that corrective surgery for tetralogy of Fallot performed in childhood is not curative surgery. Rather, this procedure only corrects the anatomic defects—without changing the progression of the disease or addressing the consequences of using a patch. As a result, these individuals develop new symptoms and eventually present to the cardiology clinic with diverse symptoms. Thus, most pediatric surgeons consider the initial surgery for tetralogy of Fallot to be long-term palliation, not definitive surgery.
With advancing age, children with tetralogy of Fallot who had corrective surgery are usually asymptomatic for the first decade after the initial procedure. Thereafter, they develop adverse myocardial problems, the majority of which involve varying degrees of pulmonary valve insufficiency, which, in turn, can lead to RV overload and RV distention and failure. [7]
RV enlargement also leads to the development of atrial and ventricular arrhythmias, which, if not recognized early, are a common cause of high morbidity and mortality. An estimated one third of adults with tetralogy of Fallot experience atrial and ventricular arrhythmias over a period of 2-3 decades. The incidence of sudden death ranges from 6% to 9% in patients who develop these arrhythmias. [8]
Another concern regarding patients with tetralogy of Fallot is that many are lost to follow-up as they become adults, and thus the opportunity to manage complications before they become irreversible can be missed. Several surveys indicate that many children with repaired tetralogy of Fallot feel fine after surgery, believe that they are cured, and then stop seeing their cardiologist. In other cases, the primary care provider is unaware of the need to follow these patients, owing to the erroneous belief that surgery is curative.
See also Tetralogy of Fallot With Pulmonary Stenosis, Tetralogy of Fallot With Pulmonary Atresia, and Tetralogy of Fallot With Absent Pulmonary Valve.
Historical information
Louis Arthur Fallot, after whom the name tetralogy of Fallot is derived, was not the first person to recognize the condition. Stensen first described it in 1672; however, it was Fallot who first accurately described the clinical and complete pathologic features of the defects.
Although the disorder was clinically diagnosed much earlier, no treatment was available until the 1940s. Cardiologist Helen Taussig recognized that cyanosis progressed and inevitably led to death in infants with tetralogy of Fallot. She postulated that the cyanosis was due to inadequate pulmonary blood flow. Her collaboration with Alfred Blalock led to the first type of palliation for these infants. In 1944, Blalock operated on an infant with tetralogy of Fallot and created the first Blalock-Taussig shunt between the subclavian artery and the pulmonary artery (see the image below).
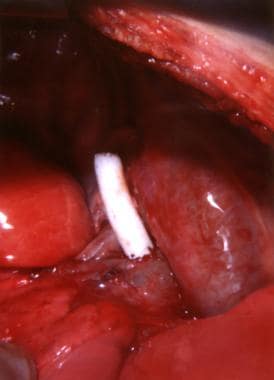
The pioneering Blalock-Taussig shunt surgical technique opened a new era in neonatal cardiac surgery. Development of the Potts shunt (from the descending aorta to the left pulmonary artery), the Glenn shunt (from the superior vena cava to the right pulmonary artery), and the Waterston shunt (from the ascending aorta to the right pulmonary artery) followed.
Scott performed the first open correction in 1954. Less than 6 months later, Lillehei performed the first successful open repair for tetralogy of Fallot using controlled cross-circulation, with another patient serving as oxygenator and blood reservoir. The following year, with the advent of cardiopulmonary bypass by Gibbons, another historic era of cardiac surgery was established. Since then, numerous advances in surgical technique and myocardial preservation have evolved in the treatment of tetralogy of Fallot.
Anatomy
The classic anatomic features of adult patients with tetralogy of Fallot (TOF) are no different from those seen in pediatric patients, such as the following [9] :
-
Perimembranous ventricular septal defect (VSD)
-
Right ventricular (RV) outflow tract obstruction (RVOTO)
-
An overriding aorta
-
RV hypertrophy
Aorta
The aorta in patients with tetralogy of Fallot is hugely dilated and overrides the diminutive pulmonary artery. In about 25% of cases, the aorta arises from a right-sided aortic arch. Because of this feature and overriding of the septum, about 50% of the blood flow from the aorta arises from the RV via the large perimembranous VSD.
RVOTO
In the majority of adults with tetralogy of Fallot, there is resistance to RV emptying. The obstruction is caused by the anterior displacement and rotation of the infundibular septum which narrows the outflow tract. The infundibular obstruction may be associated with pulmonary valve stenosis or atresia, which results in further outflow tract obstruction.
Pulmonary arteries
The size of the pulmonary arteries in tetralogy of Fallot vary in size and distribution. In most cases, the vessels are hypoplastic but can be atretic in severe cases. An almost universally common feature of tetralogy of Fallot is reduced blood flow in the pulmonary arteries and, hence cyanosis. There are also reported cases of an absent left pulmonary artery. In close to three quarters of patients with TOF, there is pulmonary valve stenosis, which is often due to leaflet tethering rather than commissural fusion. In virtually all such cases, the pulmonary annulus is narrowed.
Associated anomalies
Associated heart defects are very common in tetralogy of Fallot. Some patients may have an atrial septal defect, often referred to as part of the pentad of Fallot. Other common defects include a patent ductus arteriosus, muscular VSD, atrioventricular septal defects, anomalous coronary arteries, anomalous pulmonary venous return, absent pulmonary valve, aortic incompetence, and aortopulmonary window. [10, 11]
Etiology and Pathophysiology
Pathophysiology in pediatric patients with tetralogy of Fallot
The cause(s) of most congenital heart diseases (CHDs) are unknown, although genetic studies suggest a multifactorial etiology. Methylene tetrahydrofolate reductase (MTHFR) gene polymorphism may be a susceptibility gene for tetralogy of Fallot (TOF). [12, 13] More recently, it appears that VEGF genetic polymorphisms, -2578C>A and -634C>G, may be associated with an increased risk for tetralogy of Fallot, whereas the risk is potentially reduced with 936C>T polymorphism. [14]
Prenatal factors associated with a higher incidence of tetralogy of Fallot include maternal rubella (or other viral illnesses) during pregnancy, poor prenatal nutrition, maternal alcohol use, maternal age older than 40 years, maternal phenylketonuria (PKU) birth defects, and diabetes. Children with Down syndrome also have a higher incidence of tetralogy of Fallot, as do infants with fetal hydantoin syndrome or fetal carbamazepine syndrome.
In addition, as one of the conotruncal malformations, tetralogy of Fallot can be associated with a spectrum of lesions known as CATCH 22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia). Cytogenetic analysis may demonstrate deletions of a segment of chromosome band 22q11 (DiGeorge critical region). Ablation of cells of the neural crest has been shown to reproduce conotruncal malformations.
These abnormalities are associated with the DiGeorge syndrome and branchial arch abnormalities.
The hemodynamics of tetralogy of Fallot depend on the degree of right ventricular (RV) outflow tract obstruction (RVOTO). The ventricular septal defect (VSD) is usually nonrestrictive, and the RV and left ventricular (LV) pressures are equalized. If the obstruction is severe, the intracardiac shunt is from right to left, and pulmonary blood flow may be markedly diminished. In this instance, blood flow may depend on the patent ductus arteriosus (PDA) or bronchial collaterals.
Pathophysiology in the adult with tetralogy of Fallot
Previously, clinicians often ordered routine echocardiograms in adult patients with tetralogy of Fallot who were followed in the clinic following surgery in infancy. At that time, it was felt that, as in cases of tricuspid regurgitation, pulmonary valve insufficiency was a relatively benign and inconsequential observed entity. Because most affected patients were not symptomatic, they were never treated. However, observation of these patients over time revealed that they began to develop severe RV dysfunction and arrhythmias. The RV dilatation from the pulmonary valve insufficiency is associated with fibrosis and severe myocardial damage, which then often lead to a decrease in exercise endurance, and the majority of patients soon develop ventricular arrhythmias. [15]
Initially most patients with chronic RV function are asymptomatic, but as the compensatory mechanisms fail and the ejection fraction decreases, symptoms start to appear. If the condition is not treated at this stage, the RV dysfunction is irreversible. The arrhythmias occur as a result of progressive dilatation and stretching of the right atrium and RV.
Arrhythmias
Atrial arrhythmias of clinical significance are chiefly of the reentry type, but they may also include atrial tachycardia and atrial fibrillation. These atrial arrhythmias occur in 10-35% of patients with repaired tetralogy of Fallot. [16]
Ventricular arrhythmias and sudden death are also known to occur in those with repaired tetralogy of Fallot. Case studies have revealed that patients who experience sudden death often have moderate to severe pulmonary valve insufficiency at the time of death. Ventricular arrhythmias tend to be less common in patients with mild pulmonary valve insufficiency.
Overall, the risk of late sudden death is many times greater in patients who survive tetralogy of Fallot surgery than in their age-matched counterparts. [17]
Epidemiology
Tetralogy of Fallot (TOF) represents approximately 7%-10% of congenital heart diseases (CHDs), [2] and it is the most common cyanotic CHD, with 0.23-0.63 cases per 1,000 births. [1] This disorder accounts for one third of all CHD in patients younger than 15 years; in adults, tetralogy of Fallot has an estimated prevalence of 1 in 3,500 to 1 in 4,300 people. [2]
In most cases, tetralogy of Fallot is sporadic and nonfamilial. The incidence in siblings of affected parents is 1-5%, and it occurs more commonly in males than in females. The disorder is associated with extracardiac anomalies such as cleft lip and palate, hypospadias, and skeletal and craniofacial abnormalities. Genetic studies indicate that in some patients with tetralogy of Fallot, there may be 22q11.2 deletion and other submicroscopic copy number alterations. [18]
Adult patients with tetralogy of Fallot currently represent a very large group of patients who underwent congenital heart surgery in early life. Although the exact number of these adults is not known, because many are lost to follow-up or have never been followed, it is estimated that over two thirds of affected children who undergo repair of tetralogy of Fallot in early childhood will reach adulthood, with one study showing 94% survival rate of 168 patients aged 16 years and older who underwent simple repair. [17] For individuals born with this condition, the 30-year survival is above 75%, provided these individuals have been clinically followed. Limited data to date reveal that adult tetralogy of Fallot is equally common in both sexes.
The majority of adult patients with repaired tetralogy of Fallot present after the second or third decade of life. Males and females appear to be equally affected with symptoms as they age. [19]
Prognosis
In the present era of cardiac surgery, children with simple forms of tetralogy of Fallot (TOF) enjoy good long-term survival with an excellent quality of life. Late outcome data suggest that most survivors are in New York Heart Association (NYHA) classification I, although maximal exercise capability is reduced in some.
About 75% infants who undergo repair during infancy will survive to reach their second to third decade of life without major consequences. However, after the first two decades of life, symptoms start to appear due to pulmonary valve regurgitation. By the fourth decade of life, most survivors are symptomatic.
Adult patients with tetralogy of Fallot who undergo surgery again are usually symptom free for 10-15 years, but by the time they reach their fifth decade, even these patients begin to have symptoms. [17] Although the second surgery reduces the rate of death, the majority of these individuals have a shorter lifespan than age-matched control subjects without a history of congenital heart disease. Adults with recalcitrant arrhythmias and right heart failure have the worst prognosis.
Sudden death from ventricular arrhythmias has been reported in 1-5% of patients at a later stage in life, and the cause remains unknown. It has been suspected that ventricular dysfunction may be the cause. One study found left ventricular longitudinal dysfunction to be associated with a greater risk of developing life-threatening arrhythmias. [20] Continued cardiac monitoring into adult life is necessary. For some time, it has been suspected that certain children may have inherited a predispostion to developing long QT syndrome. A 2012 study by Chiu confirmed this suspicion. [21]
Most individuals who survive to age 30 years develop congestive heart failure (CHF), although individuals whose shunts produce minimal hemodynamic compromise have been noted, albeit rarely, and these individuals achieve a normal life span. Survival of patients into their 80s have been reported. Due to advanced surgical techniques, a 40% reduction in deaths associated with tetralogy of Fallot was noted from 1979 to 2005. [19]
Complications
Adults with repaired tetralogy of Fallot during infancy can present with the following complications later in life:
-
Right heart failure
-
Atrial and ventricular arrhythmias
-
Sudden death
-
Endocarditis
-
Septic central emboli
See Surgical Complications in patients with repaired tetralogy of Fallot.
-
Anatomic findings in tetralogy of Fallot are depicted.
-
This is a typical preoperative electrocardiogram (ECG) for tetralogy of Fallot.
-
Typical findings on postoperative electrocardiogram (ECG) for tetralogy of Fallot are shown.
-
An uplifted apex and absence of pulmonary artery segment typifies the "coeur en sabot" (ie, boot-shaped heart) of tetralogy of Fallot.
-
This angiogram shows a catheter in the right ventricle—severe infundibular stenosis.
-
This image shows completed blocking with a Taussig shunt.
-
An opening in the right ventricle exposes the ventricular septal defect.
-
Interrupted pledgetted sutures are used to close a ventricular septal defect.
-
This image shows a closed ventricular septal defect and closure of right ventriculotomy with Gore-Tex.
-
Gore-Tex is used for complete closure of right ventriculotomy.
-
Melody pulmonary valve.





