Practice Essentials
Sudden cardiac death (SCD) is an unexpected death due to cardiac causes that occurs in a short time period (generally within 1 hour of symptom onset) in a person with known or unknown cardiac disease. It is estimated that more than 7 million lives per year are lost to SCD worldwide, including over 300,000 in the United States. See the image below.
Signs and symptoms
Patients at risk for SCD may have prodromes of chest pain, fatigue, palpitations, and other nonspecific complaints. Factors relating to the development of coronary artery disease (CAD) and, subsequently, myocardial infarction (MI) and ischemic cardiomyopathy include the following:
-
Family history of premature coronary artery disease
-
Smoking
-
Dyslipidemia
-
Hypertension
-
Diabetes
-
Obesity
-
Sedentary lifestyle
Specific factors relating to cardiovascular disease are listed below.
Coronary artery disease
-
Previous cardiac arrest
-
Syncope
-
Prior myocardial infarction, especially within 6 months
-
Ejection fraction of less than 30-35%
-
History of frequent ventricular ectopy: More than 10 premature ventricular contractions (PVCs) per hour or nonsustained ventricular tachycardia (VT)
Dilated cardiomyopathy
-
Previous cardiac arrest
-
Syncope
-
Ejection fraction of less than 30-35%
-
Use of inotropic medications
-
Ventricular arrhythmias
Hypertrophic cardiomyopathy
-
Previous cardiac arrest
-
Syncope
-
Family history of SCD
-
Symptoms of heart failure
-
Drop in systolic blood pressure (SBP) or ventricular ectopy upon stress testing
-
Palpitations
-
Presence of ventricular arrhythmias
-
Considerable structural abnormality, usually defined as >3 cm left ventricular septal thickness
-
Presence of myocardial fibrosis detected by late-gadolinium enhancement in cardiac MRI
-
Most persons are asymptomatic
Valvular disease
-
Valve replacement within past 6 months
-
Syncope
-
History of frequent ventricular ectopy
-
Symptoms associated with severe, uncorrected aortic stenosis or mitral stenosis
-
Presence of bradycardia
Long QT syndrome
-
Family history of long QT and SCD
-
Medications that prolong the QT interval
-
Bilateral deafness
-
Excess QT length; usually QTc > 500 ms
See Presentation for more detail.
Diagnosis
Laboratory studies
-
Cardiac enzymes (creatine kinase, myoglobin, troponin)
-
Electrolytes, calcium, and magnesium
-
Quantitative drug levels (quinidine, procainamide, tricyclic antidepressants, digoxin): High or low drug levels may have a proarrhythmic effect
-
Toxicology screen: For drugs, such as cocaine, that cause vasospasm-induced ischemia
-
Thyroid-stimulating hormone
-
Brain natriuretic peptide (BNP)
Other tests to evaluate or predict risk of SCD in certain cases
-
Imaging studies: Chest radiography, echocardiography, nuclear scintigraphy, and/or cardiac MRI to detect the presence and extend of cardiac structual abnormality
-
Electrocardiography (ECG): Including, possibly, signal-averaged ECG to detect certain ECG signs of syndromes associated with SCD and to detect arrhythmia
-
Coronary angiography: To detect significant coronary artery disease
-
Electrophysiology study: To assess the electrical properties of the heart and the inducibility of ventricular arrhythmias; also to detect scar and fibrosis by obtaining voltage map of the heart
See Workup for more detail.
Management
In general, advanced cardiac life support (ACLS) guidelines should be followed in all cases of sudden cardiac arrest (SCA).
Bystander cardiopulmonary resuscitation (CPR)
Immediate chest compression and defibrillation are reportedly the most important interventions to improve the outcome in SCA. Research indicates that bystander use of automated external defibrillators for shockable rhythm increases neurologically intact survival to discharge (14.3% without bystander defibrillation; 49.6% with defibrillation). [1]
Pharmacologic therapy
Medications used in SCD include the following:
-
Ventricular arrhythmia: Epinephrine or vasopressin; amiodarone and lidocaine can be used as antiarrhythmic drugs if defibrillation does not control the arrhythmia. In certain cases, such as those with early repolarization syndrome or those with Brugada syndrome; VT storm should be managed by infusion of isoprotrenol. In certain cases, such as those with polymorphic VT and VF due to long QT interval, lidocaine is the preferred drug; amiodarone may prolong the QT interval and be further proarrhythmic.
-
Pulseless electrical activity (PEA): Epinephrine; atropine is used in certain case of bradycardia.
-
Asystole: One study suggested that vasopressin is more effective in acute therapy for asystole than epinephrine [2] . Both agents can be used.
-
Medical stabilization: Treat any known underlying cardiac, pulmonary, or renal problem. Empiric beta blockers are reasonable in many circumstances if the patient's hemodynamic parameters are relatively stable.
Therapeutic hypothermia
This intervention limits neurologic injury associated with brain ischemia during a cardiac arrest and reperfusion injury associated with resuscitation. [3]
Surgery
-
Temporary cardiac pacing in the case of bradycardia and bradycardia-induced VT/VF
-
Radiofrequency ablation in the case of VT storm and repeated shocks despite all medical treatments. The patient should be otherwise stable enough to tolerate the procedure.
-
Cardioverter defibrillator therapy: ICD is a very effective therapy; however, it is contraindicated in the case of VT storm. It is used more for longer term protection of the patient against future possible events. External defibrillation is the first-line therapy for witnessed or in-house VT/VF.
-
Coronary artery bypass grafting (CABG) in the case when the cause of SCD is thought to be multivessel coronary artery disease not suitable for percutaneous intervention.
-
Excision of ventricular tachycardia foci: Rarely used these days.
-
Excision of left ventricular aneurysms: Occasionally used when there is LV aneurysm with substrate for causing VT/VF that is not treatable with catheter ablation.
-
Aortic valve replacement in cases of sudden cardiac arrest (SCA) and severe aortic stenosis.
-
Ventricular assisted devices and orthotopic heart transplantation: These are usually the last measures when SCD and VT/VF fail to respond to any therapy. Not all patients are suitable for these therapies, and there are limited centers performing these procedures.
See Treatment for more detail.
Background
Sudden cardiac death (SCD) is an unexpected death due to cardiac causes occurring in a short time period (generally within 1 h of symptom onset) in a person with known or unknown cardiac disease. Most cases of SCD are related to cardiac arrhythmias. Approximately half of all cardiac deaths can be classified as SCDs. SCD represents the first expression of cardiac disease in many individuals who experience out-of-hospital cardiac arrest.
This article explores the epidemiology and pathophysiology of SCD. It also discusses the diagnostic approach to patients at risk for SCD, as well as the prevention of SCD and the treatment of sudden cardiac arrest.
For patient education information, see the Heart Health Center and Healthy Living Center, as well as Chest Pain, Arrhythmias (Heart Rhythm Disorders), Heart Disease, Heart Attack, and Cardiopulmonary Resuscitation (CPR).
Pathophysiology
The most common electrophysiologic mechanisms leading to sudden cardiac death (SCD) are tachyarrhythmias such as ventricular fibrillation (VF) or ventricular tachycardia (VT). Interruption of tachyarrhythmias, using either an automatic external defibrillator (AED) or an implantable cardioverter defibrillator (ICD), has been shown to be an effective treatment for VF and VT. [4] The implantable defibrillator has become the central therapeutic factor in the prevention and treatment of sudden cardiac death. Among the causes of SCD, ventricular tachyarrhythmias carry the best overall prognosis due to the effective treatment with defibrillation, if available.
There are multiple factors at the organ (eg imbalance of autonomic tone), tissue (eg reentry, wave break, and action potential duration alternans), cellular (eg triggered activity, and automaticity) and subcellular (abnormal activation or deactivation of ion channels) level involved in generation of VT or VF in different conditions. An anatomical or a functional block in the course of impulse propagation may create a circuit with the wave front circling around it and resulting in VT. Other mechanisms such as wave break and collisions are involved in generating VF from VT. While at the tissue level the above-mentioned reentry and wave break mechanisms are the most important known mechanisms of VT and VF, at the cellular level increased excitation or decreased repolarization reserve of cardiomyocytes may result in ectopic activity (eg automaticity, triggered activity), contributing to VT and VF initiation.
At the subcellular level, altered intracellular Ca2+ currents, altered intracellular K+ currents (especially in ischemia), or mutations resulting in dysfunction of a sodium channel (Na+ channelopathy) can increase the likelihood of VT and VF.
The balance in the interaction of all the above mentioned components of rhythm control is the key to prevent arrhythmias such as VT/VF. Those interactions are complex and involve multiple components (please see the below figure) and that is why treatment and cure of complex arrhythmias such as VT/VF have been a difficult medical and scientific task.
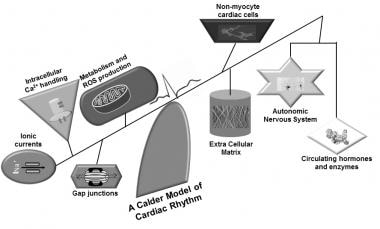 Sudden Cardiac Death. Complex arrhythmias such as ventricular tachycardia/ventricular fibrillation (VT/V)F are the result of an imbalance between important components that control the rhythm. Reprinted with permission from Nova Science Publishers, Inc (Dudley SC, Kocheril AG, Sovari AA, Eds. Ventricular Arrhythmia: From Principles to Patients, Part of the Cardiology Research and Clinical Developments Series. Nova Science Publishers. New York, 2013).
Sudden Cardiac Death. Complex arrhythmias such as ventricular tachycardia/ventricular fibrillation (VT/V)F are the result of an imbalance between important components that control the rhythm. Reprinted with permission from Nova Science Publishers, Inc (Dudley SC, Kocheril AG, Sovari AA, Eds. Ventricular Arrhythmia: From Principles to Patients, Part of the Cardiology Research and Clinical Developments Series. Nova Science Publishers. New York, 2013).
Approximately 20-30% of patients with documented sudden death events have bradyarrhythmia or asystole at the time of initial contact. Oftentimes, it is difficult to determine with certainty the initiating event in a patient presenting with a bradyarrhythmia because asystole and pulseless electrical activity (PEA) may result from a sustained VT. Less commonly, an initial bradyarrhythmia producing myocardial ischemia may then provoke VT or VF (bradycardia-dependent VT/VF).
Most cases of SCD occur in patients with structural abnormalities of the heart. Myocardial infarction (MI) and post-MI remodeling of the heart is the most common structural abnormality in patients with SCD. In patients who survive a myocardial infarction, the presence of premature ventricular contractions (PVCs), particularly complex forms such as multiform PVCs, short coupling intervals (R-on-T phenomenon), or VT (salvos of 3 or more ectopic beats), reflect an increased risk of sudden death. However suppression of the PVCs with antiarrhythmic drugs increases mortality, owing to the proarrhythmic risk of currently available medications.
Hypertrophic cardiomyopathy and dilated cardiomyopathy are associated with an increased risk of SCD. Various valvular diseases such as aortic stenosis are associated with increased risk of SCD. Acute illnesses, such as myocarditis, may provide both an initial and sustained risk of SCD due to inflammation and fibrosis of the myocardium.
Less commonly, SCD happens in patients who may not have apparent structural heart disease. These conditions are usually inherited arrhythmia syndromes. The etiology of SCD resultng from VT/VF may vary depending on the patient's age. Although coronary artery disease and ischemic cardiomyopathy are among the most common causes of VT/VF in older patients, in young patients genetic cardiomyopathies (eg hypertrophic cardiomyopathy and right ventricular cardiomyopathy) and inherited channelopathies (eg long QT syndrome and Brugada syndrome) are the most common causes.
Even though many patients have anatomic and functional cardiac substrates that predispose them to develop ventricular arrhythmias, only a small percentage develop SCD. Identifying the patients at risk for SCD remains a challenge. The strongest known predictor of SCD is significant left ventricular dysfunction of any cause. The interplay between the regional ischemia, LV dysfunction, and transient insulting events (eg, worsened ischemia, acidosis, hypoxemia, wall tension, drugs, metabolic disturbances) has been proposed as being the precipitator of sudden death (see the image below).
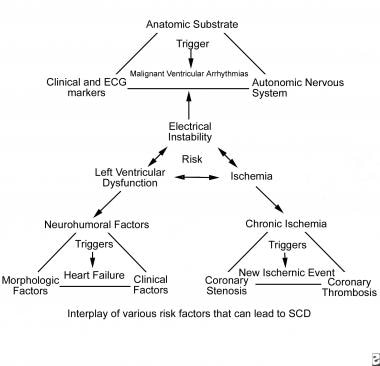 Sudden Cardiac Death. Interplay of various risk factors that can lead to sudden cardiac death.
Sudden Cardiac Death. Interplay of various risk factors that can lead to sudden cardiac death.
Etiology
A multinational group developed and validated models to predict sudden cardiac death (SCD) and pump failure death in patients with heart failure and preserved ejection fraction (HFpEF) by using data from 4116 patients in the Irbesartan in Heart Failure with Preserved Ejection Fraction trial (I-Preserve) and validating them in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM)-Preserved and Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) trials. [5] In their model, higher SCD risk was associated with the following [5] :
-
Advanced age
-
Male sex
-
Lower left ventricular ejection fraction (LVEF)
-
History of diabetes or previous myocardial infarction (MI)
-
Hospitalization for heart failure within past 6 months
Pump failure deth was predicted by the following [5] :
-
Advanced age
-
Male sex
-
Lower LVEF or diastolic blood pressure
-
Higher heart rate
-
History of diabetes or atrial fibrillation
Dyslipidemia conferred a lower risk of pump failure death. [5] The performance of both models was improved with N-terminal (NT)-pro hormone B-type natriuretic peptide (NT-proBNP) data were included.
Ischemic heart disease
Cardiac arrest due to ventricular arrhythmias may be due to post-MI remodeling of the heart with scar formation and interstitial fibrosis (intramyocardial collagen deposition) or to acute MI/ischemia. A chronic infarct scar can serve as the focus for reentrant ventricular tachyarrhythmias. This can occur shortly after the infarct or years later. Interestingly, post-MI remodeling and ischemic cardiomyopathy may be associated with increased interstitial fibrosis even in noninfarcted areas of the heart. [6] Interstitial fibrosis can provide anatomical block similar to a scar. Fibroblasts and myocytes shown to be coupled through gap junctions and fibroblasts can reduce repolarization reserve of myocytes. In addition to post-MI remodeling, many other structural heart diseases associated with sudden cardiac death (SCD) (eg, dilated cardiomyopathy, hypertrophic cardiomyopathy, and aortic stenosis) are also associated with increased myocardial fibrosis. [7, 8, 9]
Many studies support the relationship of symptomatic and asymptomatic ischemia as a factor for risk of SCD. Patients resuscitated from out-of-hospital cardiac arrest represent a group of patients with increased recurrence of cardiac arrest and have been shown to express an increased incidence of silent ST-segment depression. Experiments inducing myocardial ischemia in animal models have a strong relationship with the development of ventricular fibrillation (VF). However, in patients with prior myocardial infarction and scarring, ventricular arrhythmias, especially ventricular tachycardia (VT), do not require an acute ischemic trigger.
In postmortem studies of people who have died from SCD, extensive atherosclerosis is a common pathologic finding. In survivors of cardiac arrest, coronary heart disease with vessels showing greater than 75% stenosis is observed in 40-86% of patients, depending on the age and sex of the population studied. Autopsy studies show similar results; in one study of 169 hearts, approximately 61% of patients died of SCD, and more than 75% stenosis in three or four vessels and similar severe lesions were present in at least two vessels in another 15% of cases. No single coronary artery lesion is associated with an increased risk for SCD. Despite these findings, only approximately 20% of SCD-related autopsies have shown evidence of a recent MI. A greater proportion of autopsies (40-70%) show evidence of a healed MI. Many of these hearts also reveal evidence of plaque fissuring, hemorrhage, and thrombosis.
The Cardiac Surgery Study (CASS) showed that improving or restoring blood flow to an ischemic myocardium decreased the risk of SCD, especially in patients with 3-vessel disease and heart failure, when compared with medical treatment over a 5-year period.
The efficacy of beta-blocking agents, such as propranolol, in decreasing sudden death mortality, especially when administered to patients who had MI with VF, VT, and high-frequency PVCs, may be due in part to the ability of beta-blockers to decrease ischemia, but they are also effective in patients with nonischemic cardiomyopathy for reduction of SCD. Beta-blockers also increase the VF threshold in ischemic animals and decrease the rate of ventricular ectopy in patients who had MI.
Reperfusion of ischemic myocardium with thrombolysis or direct percutaneous coronary intervention can induce transient electrical instability by several different mechanisms.
Coronary artery spasm is a condition that exposes the myocardium to both ischemia and reperfusion insults. It is occasionally associated with VT, VF, and SCD. Since some of the episodes of coronary vasospasm may be silent, this disease should be considered in a patient with unexplained SCA. [10] The exact mechanism of ventricular arrhythmia in coronary vasospasm is not known, but factors associated with both ischemia and reperfusion may contribute in induction of arrhythmia.
Nonatherosclerotic coronary artery abnormalities, including congenital lesions, coronary artery embolism, coronary arteritis, and mechanical abnormalities of the coronary artery, have been associated with an increased incidence of sudden death.
Nonischemic cardiomyopathies
Patients with nonischemic cardiomyopathies represent the second largest group of patients who experience SCD in the United States. Nonischemic myopathies, for the purpose of this article, can be divided into the categories dilated and hypertrophic.
Dilated cardiomyopathy can result from prior ischemia and myocardial infarction or from nonischemic causes. Nonischemic dilated cardiomyopathy (DCM) is becoming increasingly more common, with an incidence of approximately 7.5 cases per 100,000 persons each year. Of cases of SCD, 10% are estimated to be attributable to DCM. The prognosis is very poor for these patients, with a 1-year mortality rate of 10-50%, depending on the New York Heart Association functional class; approximately 30-50% of these deaths are SCD.
The causes of DCM are uncertain; viral, autoimmune, genetic, and environmental (alcohol) origins are implicated. The predominant mechanism of death appears to be ventricular tachyarrhythmia, although bradyarrhythmia and electromechanical dissociation also have been observed, especially in patients with advanced LV dysfunction. Extensive fibrosis of the subendocardium, leading to dilated ventricles and subsequent generation of reentrant tachyarrhythmias, is a proposed factor in mechanism of sudden death. Multiple factors have been shown to contribute to increased risk for SCD in this population. The most important hemodynamic predictor is an increase in end-diastolic pressure and subsequent wall tension. Other important factors are increased sympathetic tone, neurohumoral activation, and electrolyte abnormalities.
Many drugs used in the treatment of heart failure, such as antiarrhythmics, inotropic agents, and diuretics, have direct or indirect (eg, through electrolyte abnormalities) proarrhythmic properties, which may provoke arrhythmias in some cases. Potassium-sparing diuretics may be helpful in decreasing SCD.
Nonsustained ventricular tachycardia (NSVT) is common in patients with dilated cardiomyopathy and approximately 80% of persons with DCM have abnormalities on Holter monitoring. Although NSVT may be a marker, it has not been shown to be a reliable predictor of SCD in these patients. Studies have shown possibility of increased mortality following suppression of NSVT by antiarrhythmic medications due to proarrhythmic properties of these medications and involvement of several other factors in generation of VT and VF. Given the possibility of sustained VT being the underlying cause, a history of syncope should be aggressively pursued. Unexplained syncope, especially in patients with class 3 or 4 heart failure, has been shown to be a predictor of SCD in most patients with cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is an autosomal-dominant, incompletely penetrant genetic disorder resulting from a mutation in one of the many (>45) genes encoding proteins of the cardiac muscle sarcomere. Among the genetic abnormalities described, mutations in the genes coding for the beta-myosin heavy chains, and cardiac troponin T make up most cases. Other mutations may include alpha-myosin heavy chain MYH6), cardiac troponin C (TNNC1), alpha-tropomyosin (TPM1), myosin binding protein-C (MYBPC3), cardiac troponin (TNNI3), essential and regulatory light-chain genes (MYL 3 and MYL 2, respectively), cardiac alpha-actin gene (ACTC), and titin (TTN). The incidence of SCD in this population is 2-4% per year in adults and 4-6% per year in children and adolescents. HCM is the most common cause of SCD in people younger than 30 years.
The vast majority of young people who die of HCM are previously asymptomatic. The patients may experience SCD while at rest or with mild exertional activity; however, in a significant portion of these patients, the SCD event occurs after vigorous exertion. HCM is the single greatest cause of SCD in young athletes and, hence, is the major entity for which to screen during the physical examination of an athlete.
The mechanism of SCD in HCM is not entirely understood. Initially, it was thought to be due to obstruction of the outflow tract because of catecholamine stimulation. However, later studies suggested that individuals with nonobstructive HCM are at high risk for SCD as well, primarily related to VT or VF. The mechanism of arrhythmia in this setting is not clear, and hypertrophy may be a part of cardiac remodeling in these patients that provides the substrate for lethal arrhythmia with a disarray of cardiac myocytes and the presence of myocardial fbrosis.
Rapid or polymorphic symptomatic NSVT may have better predictive value compared with asymptomatic and monomorphic NSVT. Other clinical markers that may have predictive value for SCD in patients with HCM are young age at onset, thickness of the septum above 3 cm, increased pressure gradiant in the outflow tract with exercise, the presence of myocardial fibrosis detected by late gadolinium enhancement in cardiac MRI, and a family history of SCD.
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic RV cardiomyopathy is characterized by replacement of the RV wall with fibrofatty tissue. Involvement of the interventricular septum and left ventricle is associated with poorer outcomes.
About 30-50% of cases occur as a phenotypically apparent familial disorder. Several genetic defects, including mutations in the desmoplakin domain locus on chromosome 6 and the ryanodine receptor locus on chromosome 1 (although this has been debated), have been correlated with SCD. Again, interstitial fibrosis plays an important role in ventricular arrhythmia in this condition. Autosomal dominant inheritance is common, but autosomal recessive transmission has been reported for select mutations. The autosomal recessive form, Naxos disease (named after the Greek Island), has been reported in a geographically isolated area mainly in Mediterranean countries and is usually associated with wooly hair and palmoplantar keratoderma or similar skin disorder. This disorder is associated with mutation in the gene for plakoglobin, a protein involved in cellular adhesion, found on chromosome 17p.
Arrhythmogenic RV dysplasia affects men more often than women. The annual incidence rate of SCD in this population is approximately 2%. Patients may present with signs and symptoms of RV hypertrophy and dilation, often with sustained monomorphic or polymorphic VT of a left bundle-branch block morphology with an axis usually between negative 90-100°
Atrial arrhythmias may be present in as many as 25% of patients. Syncope and sudden death often are associated with exercise. In many patients, sudden death is the first manifestation of the disease. Clinicians should be alerted to the epsilon wave finding on ECG studies (see the image below). The epsilon wave can be present in as many as 23% of patients after the first VT event. The percentage of patients with the epsilon wave finding on ECG increases to 27% and 34% at 5 and 10 years, respectively, after the first VT event. Another ECG sign of this condition is T wave inversion in the anterior precordial leads.
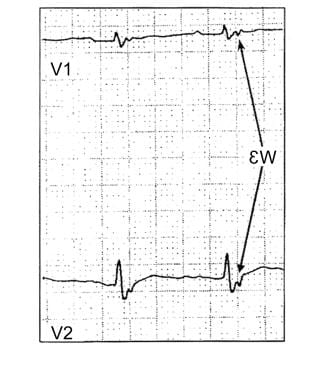 Sudden Cardiac Death. Epsilon wave in a patient with arrhythmogenic right ventricular dysplasia.
Sudden Cardiac Death. Epsilon wave in a patient with arrhythmogenic right ventricular dysplasia.
Uhl anomaly is a condition in which the RV wall is extremely thin secondary to apposition of endocardial and epicardial layers.
Valvular disease
Prior to the advent of surgical therapy for valvular heart disease, SCD was fairly common in patients with progressive aortic stenosis.
Most aortic stenosis deaths were sudden. In a study by Chizner et al of 42 patients who had isolated aortic stenosis and did not undergo valve replacement, as many as 56% of deaths were sudden at 5 years of follow-up. Of these 42 patients, 32 were symptomatic and 10 were asymptomatic. [11]
The mechanism of sudden death is unclear, and both malignant ventricular arrhythmia and bradyarrhythmia have been documented.
The incidence of SCD has decreased significantly with advent of aortic valve replacement. However, it still accounts for the second most common cause of death postoperatively in this population and especially in those with prosthetic and heterograft aortic valve replacement. The incidence of SCD after aortic valve surgery is highest in the first 3 weeks after the procedure and then plateaus at 6 months of follow-up. Transcatheter aortic valve replacement (TAVR) is a percutanous approach to replace the aortic valve. Post TAVR, certain patients may develop complete heart block and require pacemaker implantation.
Other valvular lesions
The risk of SCD is much lower in other valvular diseases compared with aortic stenosis.
Aortic insufficiency usually presents with signs of heart failure and progressive LV dilatation. As part of this process, reentrant or automatic ventricular foci may develop and ultimately lead to a symptomatic ventricular arrhythmia. After valve replacement, LV wall tension can be expected to reduce and the risk of arrhythmia can be expected to decrease.
Mitral stenosis is becoming increasingly uncommon in the United States because of widespread use of antibiotics in primary streptococcal infections. SCD due to mitral stenosis is very rare.
The incidence of SCD is low in patients with mitral valve prolapse (MVP). MVP has a 5-7% incidence in the general population. In clinically significant MVP, the risk of SCD seems to rise along with total mortality. Kligfield et al estimated that the incidence of sudden death varies with the presence of symptoms and the severity of mitral regurgitation. Ventricular tachyarrhythmias are the most frequent arrhythmia in patients with SCD. Risk factors for SCD to consider in these patients include a family history of SCD, echocardiographic evidence of a redundant mitral valve, repolarization abnormalities, and lengthening of the corrected QT interval (>420 ms in women and >450 ms in men).
Congenital heart disease
In the pediatric and adolescent age groups, SCD occurs with an incidence of 1.3-8.5 cases per 100,000 patients annually, accounting for approximately 5% of all deaths in this group. The causes of SCD are much more diverse in children than adults. In reviewing 13 studies involving 61 children and adolescents with SCD, Driscoll found 50% of cases were due to HCM; 25% were due to anomalous origin of the left coronary artery; and the remaining patients had aortic stenosis, cystic medial necrosis, and sinus node artery obstruction. The following is a classification of SCD in the pediatric population.
In patients with known, previously recognized (including repaired) congenital heart disease, abnormalities associated with SCD include the following:
-
Transposition of the great arteries
-
Fontan operation
-
Aortic stenosis
-
Hypoplastic left heart syndrome
-
Congenital heart block
In patients with known, previously recognized (including repaired) heart disease, acquired causes of SCD include the following:
-
Kawasaki syndrome
-
DCM or myocarditis
In patients with previously unrecognized heart disease who have structural heart disease, causes of SCD include the following:
-
HCM
-
Congenital coronary artery abnormalities
-
Arrhythmogenic RV cardiomyopathy
In patients with previously unrecognized heart disease who do not have structural heart disease, causes of SCD include the following:
-
Primary ventricular tachycardia and ventricular fibrillation
-
Commotio cordis - Traumatic blow to the chest wall (eg, from a hockey puck or baseball) causing VT/VF and SCD in the absence of significant identifiable trauma
The predominant mechanism is ventricular arrhythmias. In tetralogy of Fallot after postoperative correction of the anomaly, as many as 10% of these patients have VT and the incidence of sudden death is 2-3%. In the Fontan procedure, ie, to correct a physiologic single ventricle, even atrial arrhythmias can cause severe hemodynamic compromise and arrhythmic death. Patients who develop secondary pulmonary hypertension (Eisenmenger syndrome), despite attempted correction of the anatomic defects, have a very poor prognosis. The terminal event may be bradycardia or VT progressing to VF.
Primary electrophysiologic abnormalities
This generally represents a group of abnormalities in which patients have no apparent structural heart disease but have a primary electrophysiologic abnormality that predisposes them to VT or VF. Some imaging techniques have detected abnormal sympathetic neural function in these patients. An ECG study can provide clues to the diagnosis; consider a familial component to these conditions. Normal early repolarization may be associated with increased SCD, though this often represents a benign finding. [12]
Results from the Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER) suggest early repolarization is present in a significant proportion of causally diagnosed and idiopathic VF. [13]
Idiopathic long QT syndrome, in which patients have a prolonged QT with a propensity to develop malignant ventricular arrhythmias, is a rare familial disorder.
Two inheritance patterns of congenital long QT syndrome have been described. The Jervell-Lange-Nielsen syndrome, associated with congenital deafness, has an autosomal-recessive pattern of inheritance. The Romano-Ward syndrome is not associated with deafness and has an autosomal dominant pattern of inheritance with variable penetration. This syndrome accounts for 90% of long QT syndrome cases. More than 200 mutations in the 10 or more genes related to long QT syndrome have been found. Among the most common are mutations of SCN5A on chromosome 3, the HERG gene on chromosome 7, and the KVLTQT1 gene on chromosome 11.
Alteration in the function of a myocellular channel protein that regulates the potassium flux during electrical repolarization is thought to be causative, though in some subsets of long QT syndrome, such as those with mutations in SCN5A (long QT3), Na channels are primarily impaired. A relationship with sympathetic nervous system imbalance also appears to exist. The prolongation that occurs makes these patients susceptible to develop a specific form of VT called torsade de pointes.
The clinical course of patients with long QT syndrome is quite variable, with some patients remaining asymptomatic while others develop torsade de pointes with syncope and sudden death. Symptoms and SCD are more common among homozygous individuals (those with two copies of the mutant allele), compared with heterozygous individuals (who have a single mutant allele). The risk of SCD is impacted by environmental factors such as hypokalemia, medications and the presence of sinus pauses. SCD in these patients also has been associated with emotional extremes, auditory auras or stimulation, and vigorous physical activity. Symptoms usually begin in childhood or adolescence.
The probability that a specific patient has congenital long QT syndrome is divided to low, intermediate, and high probability based on the following criteria: (1) ECG criteria including long QT, torsade de pointes, notched T wave, T wave alternans, bradycardia for age; (2) clinical criteria including syncope with or without stress, deafness; and (3) family history of long QT syndrome or SCD.
When measuring QTc, selecting rhythm strips that have minimal variability of RR intervals and a stable heart rate is important.
Treatment for long QT syndrome includes beta-blockers and often pacemaker or ICD implantation. Beta-blockers decrease the overall mortality in patients with long QT syndrome. However, they do not eliminate the risk of syncope, cardiac arrest, and SCD completely. They are not effective in patients with mutation in Na channel genes (long QT3). Torsade de pointes in patients with long QT syndrome is associated with bradycardia and pauses. Therefore, a pacemaker can prevent torsade de pointes in these patients by preventing bradycardia. ICD therapy may be indicated in patients with recurrent symptoms despite treatment with beta-blockers.
Acquired long QT syndrome
A number of antiarrhythmics (especially class Ia and class III) and other medications, electrolyte abnormalities, cerebrovascular diseases, and altered nutritional states are known to cause QT prolongation and put patients at risk for torsade de pointes. This usually occurs when QT prolongation is associated with a slow heart rate and hypokalemia.
The QT interval is prolonged in as many as 32% of patients with intracranial hemorrhage (especially in subarachnoid hemorrhages). Lesions in the hypothalamus are thought to lead to this phenomenon.
Reports of sudden death due to ventricular arrhythmia in patients with hypocalcemia, hypothyroidism, nutritional deficiencies associated with modified starvation diets, and in patients who are obese and on severe weight-loss programs have been reported.
Class Ia antiarrhythmic drugs that cause acquired long QT syndrome include quinidine, disopyramide, and procainamide. Class III antiarrhythmic drugs that cause acquired long QT syndrome include sotalol, N -acetyl procainamide, bretylium, amiodarone, and ibutilide.
Other drugs that cause acquired long QT syndrome include bepridil, probucol, tricyclic and tetracyclic antidepressants, phenothiazines, Haldol, antihistamines (eg, terfenadine, astemizole), antibiotics (eg, erythromycin, sulfamethoxazole/trimethoprim), chemotherapeutics (eg, pentamidine, anthracycline), serotonin antagonists (eg, ketanserin, zimeldine), and organophosphorus insecticides.
Electrolyte abnormalities that cause acquired long QT syndrome include hypokalemia, hypomagnesemia, and hypocalcemia.
Altered nutritional states and cerebrovascular disease that cause acquired long QT syndrome include intracranial and subarachnoid hemorrhages, stroke, and intracranial trauma.
Hypothyroidism and altered autonomic status (eg, diabetic neuropathy) can cause acquired long QT syndrome.
Hypothermia can cause acquired QT prolongation. The ECG will typically also demonstrate an Osborn wave, a distinct bulging of the J point at the beginning of the ST segment. This ECG finding resolves upon warming.
Short QT syndrome
The short QT syndrome is a newly recognized syndrome, first time described in 2000, which can lead to lethal arrhythmias and SCD. Three mutations in potassium channels have been described that lead to gain of function in potassium channels and shortening of action potential and decreased QT interval.
To diagnose short QT syndrome, the QTc should be less than 330 msec and tall and peaked T waves should be present. Clinical manifestations are variable from no symptoms, to palpitations due to atrial fibrillation, syncope due to VT, and SCD. VF is easily inducible at electrophysiology study in these patients, and SCD can happen at any age.
A study by Gollob et al proposes diagnostic criteria that include QTc interval, J point–to–T peak interval, clinical history (eg, SCA, atrial fibrillation [AF], syncope), family history and genotyping. [14]
Although antiarrhythmic medications, such as sotalol, ibutilide, and procainamide, have been proposed as a therapy (to prolong the QT), data to support this approach are insufficient at present. ICD placement may be considered to prevent VT and SCD, although T-wave oversensing, resulting in inappropriate ICD discharges, has been problematic.
Because no long-term outcome data are available, Giustetto et al investigated the clinical characteristics and the long-term course of a large cohort of patients with short QT syndrome (defined as QT of ≤360 ms). Their findings suggest short QT syndrome carries a high risk of sudden death in all age groups, with the highest risk in symptomatic patients. Hydroquinidine therapy appeared to reduce the antiarrhythmic event rate from 4.9% 0%. However, this was a small registry studying a rare disease; thus, the true benefit is unclear. [15]
Wolff-Parkinson-White syndrome
WPW syndrome is a recognized but rare cause of sudden death. The existence of an atrioventricular accessory pathway in this syndrome results in ventricular preexcitation, which appears with short PR interval, wide QRS complex, and delta wave on ECG. The refractory period in the anterograde direction of accessory pathway determines the ventricular rate in the setting of atrial fibrillation and WPW. Most patients with WPW syndrome and SCD develop atrial fibrillation with a rapid ventricular response over the accessory pathway, which induces VF (see the image below). In a study by Klein et al of 31 patients with VF and WPW syndrome, a history of atrial fibrillation or reciprocating tachycardia was an important predisposing factor. The presence of multiple accessory pathways, posteroseptal accessory pathways, and a preexcited R-R interval of less than 220 ms during atrial fibrillation are associated with higher risk for SCD.
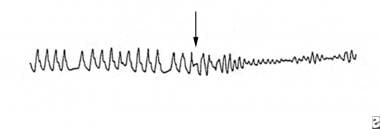 Sudden Cardiac Death. Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
Sudden Cardiac Death. Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
Symptomatic patients should be treated by antiarrhythmic medications (eg, procainamide), catheter ablation of the accessory pathway, or electrical cardioversion depending on the severity and frequency of symptoms. Asymptomatic patients may be observed without treatment.
Medications such as digoxin, adenosine, and verapamil that block the AV node are contraindicated in patients with WPW and atrial fibrillation because they may accelerate conduction through the accessory pathway, potentially causing VF and SCD.
In 1992, Brugada and Brugada described a syndrome of a specific ECG pattern of right bundle-branch block and ST-segment elevation in leads V1 through V3 without any structural abnormality of the heart, that was associated with sudden death.
In 25-30% of these patients, a mutation in SCN5A on chromosome 3 is detected. This mutation results in a sodium channelopathy. The most common clinical presentation is syncope, and this mutation is most common in young males and in Asians. It is associated with VT, VF, and SCD.
Three ECG types of Brugada pattern are described. Only type 1,- which consists of a coving ST elevation in V1 to V3 with downsloping ST segment and inverted T waves, pseudo RBBB pattern with no reciprocal ST changes and normal QTc, is specific enough to be diagnostic for Brugada syndrome when it is associated with symptoms. The other two ECG patterns of Brugada are not diagnostic, but they merit further evaluation.
The Brugada ECG pattern can be dynamic and not found on an index ECG. When clinical suspicion is high, a challenge test with procainamide or some other Na channel blocker may be diagnostic by reproducing the type 1 ECG pattern.
Although antiarrhythmic medications, catheter ablation and pacemaker therapies all have potential, in young and symptomatic patients, an ICD should be implanted to prevent VF and SCD. ICD therapy is the only proven treatment to date. Whether ICD placement is indicated in older or asymptomatic patients is controversial at present.
A prospective study by Delise et al suggests using a combination of clinical risk factors (syncope and family history of SCD) with VT inducibility in EP study to risk stratify patients with the type 1 ECG pattern of Brugada syndrome. [16]
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a syndrome that presents with polymorphic VT, syncope, or SCD, and in about half of these patients, a mutation in one of two different genes have been detected.
The polymorphic VT is characteristically induced by emotional or physical stress (eg, exercise stress test). The medical therapy of choice is administration of beta-blockers, and ICD may be indicated. New data may support the use of flecainide in the treatment of this disease. [17]
Primary ventricular fibrillation occurs in a structurally normal heart due to idiopathic etiology.
An estimated 3-9% of cases of VT and VF occur in the absence of myocardial ischemia. As many as 1% of patients with out-of-hospital cardiac arrest have idiopathic VF with no structural heart disease. As many as 15% of patients younger than 40 years who experience VF have no underlying structural heart disease. Viskin and Behassan noted that of 54 patients with idiopathic VF, 11 patients had histologic abnormalities on endomyocardial biopsy.
SCD is often the first presentation of VF in patients at risk but who have had no preceding symptoms. In those patients who survive, VF may recur in as many as one third of patients.
The options for medical therapy include beta-blockers and class 1A antiarrhythmic drugs, but limited data are available regarding their efficacy. The mainstay of treatment is preventing VF by ICD placement. Mapping and radiofrequency ablation of the triggering foci is an option for those patients who experience frequent episodes of VF following ICD placement.
Right ventricular outflow tract (RVOT) tachycardia is the most common form of idiopathic VT, comprising 70-80% of all idiopathic VTs. RVOT tachycardia is a very rare cause of SCD. It also has been referred to as exercise-induced VT, adenosine-sensitive VT, and repetitive monomorphic VT.
RVOT tachycardia occurs in patients without structural heart disease and arises from the RV outflow region. Current data suggest that triggered activity is the underlying mechanism of RVOT tachycardia. RVOT tachycardia is believed to be receptor-mediated because exogenous and endogenous adenosine can terminate this process. Maneuvers that increase endogenous acetylcholine also have been demonstrated to antagonize this process.
Symptoms typical of RVOT tachycardia include palpitations and presyncope or syncope, often occurring during or after exercise or emotional stress. VT also can occur at rest. The ECG during VT displays a left bundle-branch block/inferior axis morphology.
Treatment is based on frequency and severity of symptoms. The first line of therapy is a beta-blocker or calcium channel blocker. Patients with symptoms not relieved by medical therapy are best treated with radiofrequency catheter ablation. Successful ablation is reported in 83-100% of cases.
Other causes of sudden death
Two major causes of sudden cardiopulmonary death deserve mention.
Pulmonary embolism is a frequent cause of sudden death in people at risk. Risk factors include previous personal or family history of deep venous thromboembolism, malignancy, hypercoagulable states, and recent mechanical trauma such as hip or knee surgery.
Aortic dissection or aneurysmal rupture is the other major cause of out-of-hospital nonarrhythmic cardiovascular death. Predisposing factors for aortic dissection include genetic deficiencies of collagen such as Marfan syndrome, Ehlers-Danlos syndrome, and aortic cystic medial necrosis.
Epidemiology
United States data
Sudden cardiac death (SCD) accounts for approximately 325,000 deaths per year in the United States; more deaths are attributable to SCD than to lung cancer, breast cancer, or acquired immunodeficiency syndrome (AIDS). This represents an incidence of 0.1-0.2% per year in the adult population. SCD is often the first expression of coronary artery disease (CAD) and is responsible for approximately 50% of deaths from CAD.
In several population-based studies, the incidence of out-of-hospital cardiac arrest has been noted as declining in the past 2 decades, but the proportion of sudden CAD deaths in the United States has not changed. A high incidence of SCD occurs among certain subgroups of high-risk patients (congestive heart failure with ejection fraction < 30%, convalescent phase after myocardial infarction, patients who survived cardiac arrest). However, these populations are much smaller than patients with minimal or even inapparent coronary artery disease. Consequently, in the overall population, most SCD occurs in lower risk patients. The time dependence of risk for SCD has been noted in several studies, with an increased number of events in the first 6-24 months after surviving a major cardiovascular event.
International data
The frequency of SCD in Western industrialized nations is similar to that in the United States. The incidence of SCD in other countries varies as a reflection of the prevalence of coronary artery disease or other high-frequency cardiomyopathies in those populations. The trend toward increasing SCD events in developing nations of the world is thought to reflect a change in dietary and lifestyle habits in these nations. It has been estimated that SCD claims more than 7,000,000 lives per year worldwide. [18]
Race-related demographics
Most studies demonstrate inconclusive data with regard to racial differences as they relate to the incidence of sudden death. Some studies suggest that a greater proportion of coronary deaths were "sudden" in blacks compared to whites. In a report by Gillum et al on SCD from 1980-1985, the percentage of coronary artery disease deaths occurring out of the hospital and in EDs was found to be higher in blacks than in whites (see the image below). [19]
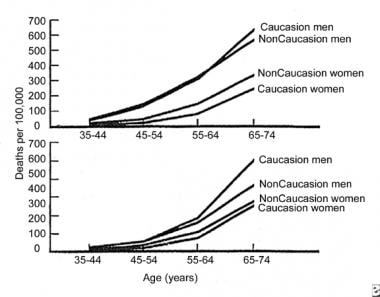 Sudden Cardiac Death. Plots of mortality rates (deaths per 1000 persons) for ischemic heart disease occurring out of the hospital or in the emergency department (top) and occurring in the hospital (bottom) by age, sex, and race in 40 states during 1985.
Sudden Cardiac Death. Plots of mortality rates (deaths per 1000 persons) for ischemic heart disease occurring out of the hospital or in the emergency department (top) and occurring in the hospital (bottom) by age, sex, and race in 40 states during 1985.
Sex-related demographics
Men have a higher incidence of SCD than women, with a ratio of 3:1. This ratio generally reflects the higher incidence of obstructive coronary artery disease in men. Relatively recent evidence suggests that a major sex difference may exist in the mechanism of myocardial infarction. Basic and observational data point to the fact that men tend to have coronary plaque rupture, while women tend to have plaque erosion. Whether this biologic difference accounts for the male predominance of SCD is unclear.
Age-related demographics
The incidence of SCD parallels the incidence of coronary artery disease, with the peak of SCD occurring in people aged 45-75 years. The incidence of SCD increases with age in men, women, whites, and nonwhites as the prevalence of coronary artery disease increases with age. However, the proportion of deaths that are sudden from coronary artery disease decreases with age. In the Framingham study, the proportion of coronary artery disease deaths that were sudden was 62% in men aged 45-54 years, but this percentage fell to 58% in men aged 55-64 years and to 42% in men aged 65-74 years. [20] According to Kuller et al, 31% of deaths are sudden in people aged 20-29 years. [21]
Prognosis
Prognosis of morbidity and mortality for people who have had sudden cardiac arrest (SCA) can be made using the cardiac arrest score developed by McCullough and Thompson (see Physical Examination). The detection of the underlying cause of sudden cardiac death (SCD) and available treatment options play an important role in the natural history and prognosis of SCD.
SCD/SCA is a frequently encountered problem for emergency physicians, internists, and cardiologists. Ischemic cardiomyopathy in all adult cases and HCM in pediatric and adolescent cases are at the top of the list of causes of SCA.
The clinical course, once the patient is resuscitated, largely is predicted by the emergency department (ED) presentation of hemodynamic stability, early neurologic recovery, and the duration of the resuscitation.
Patients who survive the initial phases require a systematic evaluation of left ventricular (LV) performance, myocardial perfusion, and electrophysiologic instability. Survivors of SCA have a recurrence rate on the order of 20-25% per year, making implantable cardioverter defibrillator (ICD) implantation important in the majority of these patients.
ICD implantation saves lives. Risk stratification will continue to be an area of active research.
Preventive measures, at their roots, are measures of coronary artery disease prevention. Efforts to inform and train the public about external defibrillator use likely will have a great public health impact on improving survival rates of SCA. However, more basic and clinical research is required to understand the mechanism of VF/VT and to be able to identify the patients at risk who benefit from ICD therapy.
Morbidity/mortality
Of more than 300,000 deaths attributed to sudden cardiac death (SCD) in the United States each year, a large portion (as many as 40%) are unwitnessed. For most people who experience SCD, their survival depends on the presence of individuals who are competent in performing basic life support, the rapid arrival of personnel and apparatus for defibrillation and advanced life support, and transfer to a hospital. Even under ideal circumstances, only an estimated 20% of patients who have out-of-hospital cardiac arrest survive to hospital discharge. In a study of out-of-hospital cardiac arrest survival in New York City, only 1.4% of patients survived to hospital discharge. Other studies in suburban and rural areas have indicated higher rates of survival (as high as 35%). Placement of automatic external defibrillators throughout communities and training people to use them has the potential to markedly improve outcomes from SCD.
-
Upon emergency department (ED) presentation, the most important determinants of survival include (1) an unsupported systolic blood pressure (SBP) greater than 90 mm Hg, (2) a time from loss of consciousness to return of spontaneous circulation (ROSC) of less than 25 minutes, and (3) some degree of neurological responsiveness.
-
A major adverse outcome from a SCD event is anoxic encephalopathy, which occurs in 30-80% of cases.
-
Sudden Cardiac Death. Interplay of various risk factors that can lead to sudden cardiac death.
-
Sudden Cardiac Death. Plots of mortality rates (deaths per 1000 persons) for ischemic heart disease occurring out of the hospital or in the emergency department (top) and occurring in the hospital (bottom) by age, sex, and race in 40 states during 1985.
-
Sudden Cardiac Death. Figure a: Neurologic outcome stratified by initial cardiac arrest score. Neurologic recovery is defined as discharged to home and able to care for self. Figure b: Overall survival stratified by initial cardiac arrest score.
-
Sudden Cardiac Death. Epsilon wave in a patient with arrhythmogenic right ventricular dysplasia.
-
Sudden Cardiac Death. Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
-
Sudden Cardiac Death. Complex arrhythmias such as ventricular tachycardia/ventricular fibrillation (VT/V)F are the result of an imbalance between important components that control the rhythm. Reprinted with permission from Nova Science Publishers, Inc (Dudley SC, Kocheril AG, Sovari AA, Eds. Ventricular Arrhythmia: From Principles to Patients, Part of the Cardiology Research and Clinical Developments Series. Nova Science Publishers. New York, 2013).





