Practice Essentials
Protein S is a vitamin K–dependent anticoagulant protein that was first discovered in Seattle, Washington in 1979 and arbitrarily named after that city. The major function of protein S is as a cofactor to facilitate the action of activated protein C (APC) on its substrates, activated factor V (FVa) and activated factor VIII (FVIIIa).
Protein S deficiency usually manifests clinically as venous thromboembolism (VTE). [1] Any association of protein S deficiency with arterial thrombosis appears coincidental or weak at best. Evidence for arterial thrombosis in other hereditary thrombophilias (eg, protein C deficiency, antithrombin III deficiency, or factor V Leiden gene mutation) also appears to be minimal. [2]
Protein S deficiency may be hereditary or acquired; the latter is usually due to hepatic disease, nephrotic sydrome, or vitamin K deficiency. Protein S and C levels are lower in sickle cell anemia and they decrease further significantly during crisis. [3] Acquired protein S deficiency has been reported in patients infected with COVID-19 and is associated with increased risk of thrombotic events in these patients. [4]
Hereditary protein S deficiency is an autosomal dominant trait. Thrombosis is observed in both heterozygous and homozygous genetic deficiencies of protein S.
Protein S deficiency is primarily diagnosed using laboratory tests that detect free protein S antigen, and less commonly by measuring functional protein S activity (based on clotting assays); see Workup. Management is required in the event of acute VTE and includes administration of a low molecular weight heparin (LMWH), a vitamin K antagonist, or a direct oral anticoagulant (DOAC). Prophylaxis may be used in selected patients who are asymptomatic carriers without a thrombotic event. (See Treatment and Medication.)
For patient education information, see the Deep Vein Thrombosis Health Center and What Is Thrombophilia?.
Pathophysiology
To understand how thrombosis occurs in protein S deficiency, its physiological function should be briefly reviewed. Protein S is part of a system of anticoagulant proteins that regulate normal coagulation mechanisms in the body. [5] Under most normal circumstances, the anticoagulant proteins prevail and blood remains in a liquid nonthrombotic state. Whenever procoagulant forces are locally activated to form a physiologic or pathologic clot, protein S participates as part of one mechanism of controlling clot formation. [6]
Protein S functions predominantly as a nonenzymatic cofactor for the action of another anticoagulant protein, activated protein C (APC). This activity occurs via a coordinated system of proteins, termed the protein C system. The image below shows a simplified outline of the function of protein S in the protein C system.
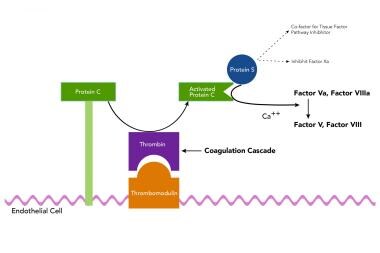 A simplified outline of the function of protein S in the protein C system.
A simplified outline of the function of protein S in the protein C system.
During the process of clotting, multimolecular complexes are formed on membrane surfaces. These membranes are usually negatively charged phospholipids and/or activated platelets. These multimolecular complexes are referred to as the tenase and prothrombinase complexes for their key activities of activation of factor X and prothrombin, respectively. Anchoring these two complexes are the activated form of factor VIII (FVIIa) used for the tenase complex and the activated form of factor V (FVa) for the prothrombinase complex. These two large proteins are homologous in structure and are cofactors, not enzymes, in the clotting process.
In one of many examples of nature's efficiency, the same enzyme that clots blood, thrombin, is converted from clotting to an anticoagulant mechanism on the surface of the endothelium and it then activates protein C to its active enzymatic form, APC. APC requires protein S as a cofactor in its enzymatic action on its 2 substrates, FVa and FVIIIa. Thus, this process is designed to dampen and shut off clotting by switching off the key cofactor proteins FVa and FVIIIa. Protein S and APC are sufficient to inactivate FVa. However, for the inactivation of FVIIIa, APC and protein S require the help of the nonactivated clotting protein, factor V. This is another example of dual use of a protein in this same process.
Factor Va as noted above is cleaved by APC to an inactive form. However, in assisting protein S to inactivate FVIIIa, it is the inactive FV that is cleaved by APC. An important consequence of this dual procoagulant and anticoagulant property of factor V, is that the mutant factor V Leiden, which resists APC cleavage, cannot be switched off but also cannot function here at this step as an anticoagulant protein (see factor V Leiden gene mutation in Race). In addition to its cofactor role in the protein C system, protein S functions independently of protein C by acting as a cofactor to Tissue Factor Pathway Inhibitor (TFPI). TFPI inhibits the Tissue Factor/FVIIa complex thus inhibiting activation of factor X as well as Prothrombin further downstream. Similarly, Protein S has also been shown to directly inhibit factor Xa. [7, 8]
Heeb et al reported that protein S has APC-independent anticoagulant activity, termed PS-direct, that directly inhibits factor Xa/factor Va prothrombinase complex, a process made possible by the presence of zinc (Zn2+) content in protein S. The investigators found Zn2+ content positively correlated with PS-direct in prothrombinase and clotting assays, but the APC-cofactor activity of protein S was independent of Zn2+ content. In addition, protein S that contained Zn2+ bound factor Xa more efficiently than protein S without Zn2+, and, independent of Zn2+ content, protein S also efficiently bound tissue factor pathway inhibitor. [9] The study also suggested that conformation differences at or near the interface of 2 laminin G-like domains near the protein S C terminus may indicate that Zn2+ is necessary for PS-direct and efficient factor Xa binding and could have a role in stabilizing protein S conformation. [9]
Protein S is a single-chain glycoprotein, and it is dependent on vitamin K action for posttranslational modification of the protein to a normal functional state. Vitamin K–dependent proteins are synthesized with a unique recognition propeptide piece. The propeptide sequence serves as a recognition site for the vitamin K–dependent gamma-carboxylase enzyme that modifies the nearby glutamic acid residues to gamma-carboxyglutamic acid (Gla) residues. Gla residues are responsible for calcium-dependent binding to membrane surfaces. Structural studies indicate that protein S contains 10-12 Gla residues, a loop region sensitive to thrombin (ie, thrombin-sensitive region [TSR]), 4 epidermal growth factor (EGF)–like modules, and a carboxy-terminal portion that is homologous to a sex hormone-binding globulin (SHBG)–like region.
In blood plasma, protein S exists in both a bound and a free state. A portion of protein S is noncovalently bound with high affinity to the complement regulatory protein C4b-binding protein (C4BP). The C4BP molecule consists of 7 alpha chains that bind to the complement protein, C4b, and one beta chain. The beta chain of the C4bBP molecules contains the binding sites for protein S. There is emerging evidence in the role of Protein S in the complement pathway. It is now found to interact with the complement system and may play a role in phagocytosis of apoptotic cells. Protein S interacts with tyrosine kinase receptors of the TAM family, along with phosphatidyl serine on cell surface apoptotic cells which stimulates macrophage phagocytosis of these cells. [10, 11] The physiological impact of protein S deficiencies on these nonanticoagulant roles of protein S is not yet known.
APC and protein S require negatively charged phospholipids (PL) and Ca2+ for normal anticoagulant activity. Studies of the structure and function relationships of protein S demonstrate that the APC interaction sites are located in the Gla, TSR, and first EGF-like modules of protein S. The binding site for C4BP is located in the SHBG-like region, which is also important for full anticoagulant activity.
In healthy individuals, approximately 30-40% of total protein S is in the free state. Only free protein S is capable of acting as a cofactor in the protein C system. This distinction between free and total protein S levels is important and gives rise to the current terminology regarding the deficiency states. Type I protein S deficiency is a reduction in the level of free and total protein S. Type III deficiency is a reduction in the level of free protein S only. Type II deficiency is a reduction in the cofactor activity of protein S, with normal antigenic levels.
Age affects total protein S but not free protein S levels. Generally, the total protein S level increases in persons older than 50 years. This rise is in association with total increases in the complement binding protein, C4BP. Free protein S levels do not increase with age. These factors may explain the observation that families with the same recognized genetic defect in protein S can have both type I and type III deficiencies. When families with the same genetic type I defect are surveyed, older individuals even with deficiency in protein S have an increase in total protein S and now appear to have type III deficiency.
Etiology
Protein S deficiency may be hereditary or acquired.
Hereditary protein S deficiency
Researchers have identified 2 genes for human protein S; both are linked closely on chromosome 3p11.1-3q11.2. [12] One gene is the active gene, PROS-α (ie, PROS1), and the other, PROS-β, is an evolutionarily duplicated nonfunctional gene, which is classified as a pseudogene because it contains multiple coding errors (eg, frameshifts, stop codons). [13] The expressed (alpha) PROS1 gene is more than 80 kb long and contains 15 exons and 14 introns. The protein S pseudogene (beta) has 97% homology to the PROS-α gene. [14] Molecular studies into the genetic causes of protein S deficiency are complicated by the presence of the pseudogene, PROS-β, and phenotypic variation.
Over 200 mutations in PROS1 have been identified as causes of protein S deficiency and thrombophilia. Most are point mutations, such as transversion mutations that generate a premature stop codon and thus result in a truncated protein S molecule. [15, 16, 17] A missense mutation in exon 7 of PROS1 in which glycine is replaced with arginine has been reported in a Chinese family. In addition, deletions of large portions of the PROS1 gene have been reported. Researchers located the first such deletion in the central portion of the gene. [18] The second deletion described (5.3 kb) was a deletion of coding exon 13, which resulted in a truncated protein product. [19]
Wu and colleagues conducted a case-control study of 603 Han Chinese patients with venous thromboembolism. Gene sequencing identified 24 different mutations in 34 patients with protein S deficiency with 50% of the mutations around exons 11 and 12 of PROS1. [20]
Acquired protein S deficiency
Acquired conditions associated with decreased protein S levels include the following:
-
Oral contraceptive use
-
Warfarin anticoagulant use
-
Nephrotic syndrome
-
Vitamin K deficiency
-
Chronic liver disease
-
Certain viral infections (eg, HIV, varicella)
-
Systemic lupus erythematosus
-
Myeloproliferative disorders
-
COVID-19
In addition, protein S levels decrease in pregnancy and can fall into the abnormal-low laboratory range. These low levels of protein S in pregnancy do not cause thrombosis by themselves.
Another seldom recognized cause for acquired protein S deficiency is sickle cell disease. However, this condition alone does not produce a thrombophilic state.
Epidemiology
Frequency
United States
Protein S deficiency is rare in the healthy population without VTE. In a study of 3788 healthy blood donors, the prevalence of famililial protein S deficiency was 0.03 to 0.13%. [21] When a selected group of patients with recurrent thrombosis or family history of thrombosis is analyzed, the frequency of protein S deficiency increases to 3-5%. [22, 23]
Studies evaluating the clinical significance of free protein S levels associated with risk of VTE suggest using a lower cutoff of protein S levels for the diagnosis, which would in turn affect the prevalance of the disease. [24] The Multi Environmental and Genetic Assessment Study (MEGA) case control study used protein S levels of less than 2.5th percentile of controls to identify protein S deficiency; however, risk of unprovoked VTE was limited to patients with free protein S levels of less than 0.10th percentile (< 33 U/dL). The prevalence of patients in this subgroup was 0.4%. [25]
International
Data for European studies indicate the same frequencies for protein S deficiency as in the United States. In contrast, the prevalence of protein S deficiency is particularly high in the Japanese population, with a frequency of approximately 12.7% in patients with VTE and approximately 0.48%-0.63% in the general population. [26] In a Japanese study that evaluated patients with VTE for congenital thrombophilia, Ikejiri et al diagnosed congenital protein S deficiency in eight of 130 patients (6.2%). [27]
Race
Race-related variations exist in thrombophilic disorders, as one would expect with genetically based traits. In general, a significant difference exists in the frequency of thrombophilic disorders in whites compared with Japanese (Asian) persons and African Americans. Current research indicates that protein S deficiency is 5-10 times more common in Japanese populations than in whites. Protein C deficiency is estimated to be 3 times higher in Japanese populations as well.
The factor V Leiden mutation is common in white populations and is now known to be the result of a founder effect estimated to be 30,000 years old. This mutation is almost never found in Japanese or Asian populations. In general, black Africans and African Americans with VTE have a lower detection rate of any of the currently recognized thrombophilic disorders, especially factor V Leiden.
Sex
Men have a higher level of both free and total protein S antigen, the clinical relevance of which is not clearly documented. [21]
Age
In hereditary protein S deficiency, the age of onset of thrombosis varies by heterozygous versus homozygous state. Most VTE events in heterozygous protein S deficiency occur in persons younger than 40-45 years. The rare homozygous patients have neonatal purpura fulminans, as described above; onset occurs in early infancy. As noted above in the discussion on genetics, age does affect total protein S antigen levels, but not free protein S levels. Older patients deficient in protein S have low free S levels, even if their total protein S level rises into the normal range.
Prognosis
Congenital protein S deficiency is an autosomal dominant disease, with variable penetrance and heterogeneous genetic basis. VTE develops in almost 50% of patients who are heterozygous for protein S deficiency. The remaining patients are asymptomatic and some heterozygous individuals never develop VTE. Annual incidence of venous thrombosis was found to be 1.90%, with median age of presentation being 29 yrs in a retrospective cohort study of 2479 relatives. [23]
Very rarely, protein S deficiency occurs as a homozygous state, and these individuals have a characteristic thrombotic disorder, purpura fulminans. Purpura fulminans is characterized by small-vessel thrombosis with cutaneous and subcutaneous necrosis, and it appears early in life, usually during the neonatal period or within the first year of life. [28]
Though it is controversial, no clear association exists between protein S deficiency and arterial thrombosis. Many case reports and small case series describe protein S deficiency as one factor in patients with arterial thrombosis, most commonly stroke. However, prospective and cohort studies have not shown convincing increased risk for arterial thrombosis.
Protein S deficiency is also associated with fetal loss in women, in the absence of VTE. Some authors suggest that as many as 40% of women with obstetric complications other than VTE may carry some form of thrombophilia. Protein S deficiency is one of these factors along with several other more common genetic thrombophilic states.
Cases of warfarin-induced skin necrosis have been reported in patients with protein S deficiency. [29]
Mortality is from pulmonary embolism. In several studies, the 3-month mortality rate of pulmonary embolism ranged from 10.0-17.5%. In a study of Medicare recipients with pulmonary embolism, men had a 13.7% mortality rate compared to 12.8% in women; the mortality rate was 16.1% in blacks, compared to 12.9% in whites. [30]
-
A simplified outline of the function of protein S in the protein C system.