Practice Essentials
Acute kidney injury (AKI) is a clinical syndrome manifested by a rapid or abrupt decline in kidney function and subsequent dysregulation of the body electrolytes and volume, and abnormal retention of nitrogenous waste. The widely accepted Kidney Disease: Improving Global Outcome (KDIGO) definition of AKI is based on the change of serum creatinine and urine output, as follows [1] :
-
Rise in serum creatinine ≥0.3 mg/dL within 48 hours
-
Rise in serum creatinine ≥1.5 times baseline, which is known or presumed to have occurred within the prior seven days
-
Urine output < 0.5 mL/kg/hour for six hours
Symptoms and signs
Most patients with AKI have no clinical symptoms related to AKI and are diagnosed on the basis of a routine laboratory blood test. Depending on the degree of kidney function impairment and the duration, however, they might have hypertension, edema, decreased urine output, shortness of breath, anorexia, nausea, sleep disturbances and altered mental status. When evaluating a patient with AKI, the signs listed below may help in identifying the etiology associated with AKI.
Skin:
-
Livedo reticularis, digital ischemia, butterfly rash, and palpable purpura: Autoimmune disease and systemic vasculitis
-
Maculopapular rash: Allergic interstitial nephritis
-
Track marks (ie, intravenous drug abuse): Endocarditis
Eyes:
-
Keratitis, iritis, uveitis, dry conjunctivae: Autoimmune vasculitis
-
Jaundice: Liver disease
-
Band keratopathy (ie, hypercalcemia): Multiple myeloma
-
Signs of diabetes mellitus
-
Signs of hypertension
-
Atheroemboli: Retinopathy (ie, Hollenhorst plaque in cholesterol microembolism)
Ears:
-
Hearing loss: Alport disease, aminoglycoside toxicity
-
Mucosal or cartilaginous ulcerations: granulomatosis with polyangiitis (previously Wegener granulomatosis)
Cardiovascular system:
-
Irregular rhythms (eg, atrial fibrillation): Thromboemboli, hypotension
-
Murmurs: Endocarditis, valvular heart disease
-
Pericardial friction rub: Uremic pericarditis
-
Increased jugular venous distention, rales, S 3: Cardiomyopathy
Abdomen:
-
Pulsatile mass or bruit: Renal artery stenosis, atheroemboli
-
Abdominal or costovertebral angle tenderness: Pyelonephritis, nephrolithiasis, papillary necrosis, renal artery thrombosis, renal vein thrombosis
-
Pelvic, rectal masses; prostatic hyperplasia; distended bladder: Urinary obstruction
Pulmonary system:
-
Rales: Pulmonary edema, pulmonary infection
-
Hemoptysis: ANCA vasculitis, anti-glomerular basement membrane (Goodpasture syndrome or anti-GBM disease)
See Presentation for more detail.
Diagnosis
The following tests can aid in the diagnosis and assessment of AKI:
-
Kidney function studies: Increased levels of blood urea nitrogen (BUN) and serum creatinine are the hallmarks of AKI; the ratio of BUN to creatinine can exceed 20:1 in conditions that favor the enhanced reabsorption of urea, such as volume contraction (this suggests prerenal AKI)
-
Complete blood count: Elevated WBC can indicate infection; risk-stratify for tumor lysis syndrome in an oncologic patient; low hemoglobin can suggest acute blood loss or chronic anemia; thrombocytopenia might indicate thrombotic microangiopathy or portal hypertension
-
Peripheral smear: Schistocytes may indicate hemolytic-uremic syndrome or thrombotic thrombocytopenic purpura)
-
Serologic tests: May show evidence of systemic diseases associated with AKI, such as lupus nephritis, ANCA vasculitis, anti-GBM disease or Goodpasture syndrome
-
Complement testing: Pattern may indicate AKI related to endocarditis or various glomerulonephritidites
-
Fractional excretion of sodium and urea in the setting of oliguria
-
Bladder pressure: Patients with a bladder pressure above 25 mm Hg should be suspected of having AKI caused by abdominal compartment syndrome
-
Ultrasonography: Renal ultrasonography is helpful in evaluating existing renal disease (kidney length, presence of cortical echogenicity or cortical thinning) and urinary obstruction
-
Aortorenal angiography: Can help establish the diagnosis of renal vascular diseases, such as renal artery stenosis, renal atheroembolic disease, atherosclerosis with aortorenal occlusion, and instances of necrotizing vasculitis (eg, polyarteritis nodosa)
-
Kidney biopsy: Can be useful in identifying intrarenal causes of AKI and directing targeted therapy
See Workup for more detail.
Management
In addition to treating the underlying etiology, maintenance of volume homeostasis and correction of biochemical abnormalities remain the primary goals of AKI treatment (supportive care) and may include the following measures:
-
Correction of fluid overload with loop diuretics and fluid restriction
-
Correction of severe acidosis with alkali administration
-
Correction of life-threatening hyperkalemia
-
Correction of hematologic abnormalities (eg, blood loss anemia, uremic platelet dysfunction) with measures such as RBC or platelet transfusions and administration of desmopressin or estrogens
-
Hemodialysis in patients with refractory acidosis, hypervolemia with pulmonary edema, life-threatening hyperkalemia, and uremic signs or symptoms
Dietary changes are an important facet of AKI treatment. Restriction of sodium, potassium, and fluids becomes crucial in the management of oliguric AKI with hyperkalemia, in which the kidneys do not adequately excrete either toxins or fluids.
Non-targeted pharmacologic interventions for AKI have been attempted, but no large randomized controlled study has demonstrated significant benefit.
See Treatment and Medication for more details.
For patient education information, see Acute Kidney Failure.
Background
Acute kidney injury (AKI) is defined as an abrupt or rapid decline in renal filtration function. This condition is usually marked by a rise in serum creatinine concentration or azotemia (a rise in blood urea nitrogen [BUN] concentration). [2] However, immediately after a kidney insult, BUN or serum creatinine levels may be normal. In the early phase, the only sign of a kidney injury may be decreased urine production. (See History.)
Furthermore, a rise in serum creatinine might not always be related to a decrease in kidney function; certain medications (eg, cimetidine, trimethoprim, Poly ADP-ribose polymerase [PARP] inhibitors, and cyclin-dependent kinase 4 and 6 [CDK4/6] inhibitors) can inhibit the kidney’s tubular secretion of creatinine independent of glomerular filtration rate (GFR). A rise in the BUN level can also occur without renal injury, as a result of gastrointestinal (GI) or mucosal bleeding, steroid use, or protein loading. Therefore, a careful inventory must be taken before concluding that a kidney injury is present. (See Etiology and History.)
See Chronic Kidney Disease and Acute Tubular Necrosis for complete information on these topics. For information on pediatric cases, see Chronic Kidney Disease in Children.
Categories of AKI
Traditionally, AKI may be classified into 3 general categories as follows:
-
Prerenal - As an adaptive response to severe volume depletion and hypotension, with structurally intact nephrons
-
Intrinsic - In response to cytotoxic, ischemic, or inflammatory insults to the kidney, with structural and functional damage
-
Postrenal - From obstruction to the passage of urine
While this classification helps guide the development of a differential diagnosis, many pathophysiologic features are shared among the different categories. (See Etiology.)
Oliguric and nonoliguric patients with AKI
Patients who develop AKI can be oliguric or nonoliguric, have a rapid or slow rise in creatinine levels and may have qualitative differences in urine solute concentrations and cellular content. (Approximately 50-60% of all causes of AKI are nonoliguric.) This lack of a uniform clinical presentation reflects the variable nature of the injury.
Classifying AKI as oliguric or nonoliguric on the basis of daily urine excretion has prognostic value. Oliguria is defined as a daily urine volume of less than 400-500 mL, which is the minimum amount of urine required to eliminate the average daily solute load and has a worse prognosis.
Anuria is defined as a urine output of less than 50-100 mL/day and, if abrupt in onset, suggests bilateral obstruction or catastrophic injury to both kidneys.
Stratification of kidney injury along these lines helps in diagnosis and decision-making (eg, timing of dialysis) and can be an important criterion for patient response to therapy.
RIFLE classification system
In 2004, the Acute Dialysis Quality Initiative workgroup set forth a definition and classification system for acute renal failure, described by the acronym RIFLE (Risk of renal dysfunction, Injury to the kidney, Failure or Loss of kidney function, and End-stage kidney disease). [3] Investigators have since applied the RIFLE system to the clinical evaluation of AKI, although it was not originally intended for that purpose. AKI research increasingly uses RIFLE. See Table 1 below.
Table 1. RIFLE Classification System for Acute Kidney Injury (Open Table in a new window)
Stage |
GFR Criteria |
Urine Output Criteria |
Probability |
Risk |
SCreat increased × 1.5 or GFR decreased >25% |
UO < 0.5 mL/kg/h × 6 h |
High sensitivity (Risk >Injury >Failure) |
Injury |
SCreat increased × 2 or GFR decreased >50% |
UO < 0.5 mL/kg/h × 12 h |
|
Failure |
SCreat increased × 3 or GFR decreased 75% or SCreat ≥4 mg/dL; acute rise ≥0.5 mg/dL |
UO < 0.3 mL/kg/h × 24 h (oliguria) or anuria × 12 h |
|
Loss |
Persistent acute renal failure: complete loss of kidney function >4 wk |
High specificity |
|
ESKD |
Complete loss of kidney function >3 mo |
||
ESKD—end-stage kidney disease; GFR—glomerular filtration rate; SCreat—serum creatinine; UO—urine output Note: Patients can be classified by GFR criteria and/or UO criteria. The criteria that support the most severe classification should be used. The superimposition of acute on chronic failure is indicated with the designation RIFLE-FC; failure is present in such cases even if the increase in SCreat is less than 3-fold, provided that the new SCreat is greater than 4.0 mg/dL (350 µmol/L) and results from an acute increase of at least 0.5 mg/dL (44 µmol/L). |
|||
When the failure classification is achieved by UO criteria, the designation of RIFLE-FO is used to denote oliguria.
The initial stage, risk, has high sensitivity; more patients will be classified in this mild category, including some who do not actually have kidney failure. Progression through the increasingly severe stages of RIFLE is marked by decreasing sensitivity and increasing specificity.
Acute Kidney Injury Network classification system
The Acute Kidney Injury Network (AKIN) has developed specific criteria for the diagnosis of AKI. The AKIN defines AKI as abrupt (within 48 hours) reduction of kidney function, after excluding urinary obstruction and achieving adequate hydration, manifested by any 1 of the following [4]
-
An absolute increase in serum creatinine of 0.3 mg/dL or greater (≥26.4 µmol/L)
-
A percentage increase in serum creatinine of 50% or greater (1.5-fold from baseline)
-
A reduction in urine output, defined as less than 0.5 mL/kg/h for more than 6 hours
AKIN has proposed a staging system for AKI that is modified from RIFLE. In this system, either serum creatinine or urine output criteria can be used to determine the stage. See Table 2 below.
Table 2. Acute Kidney Injury Network Classification/Staging System for AKI (Open Table in a new window)
Stage |
Serum Creatinine Criteria |
Urine Output Criteria |
1 |
Increase of ≥0.3 mg/dL (≥26.4 µmol/L) or 1.5- to 2-fold increase from baseline |
< 0.5 mL/kg/h for >6 h |
2 |
> 2-fold to 3-fold increase from baseline |
< 0.5 mL/kg/h for >12 h |
3* |
> 3-fold increase from baseline, or increase of ≥ 4.0 mg/dL (≥35.4 µmol/L) with an acute increase of at least 0.5 mg/dL (44 µmol/L) |
< 0.3 mL/kg/h for 24 h or anuria for 12 h |
*Patients who receive renal replacement therapy (RRT) are considered to have met the criteria for stage 3 irrespective of the stage they are in at the time of RRT. |
||
KDIGO classification system
The KDIGO system, which is the most recent and widely accepted classification, was developed by merging the RIFLE and AKIN classifications into a single simplified one. It offers equivalent or superior sensitivity for AKI detection and prognostic performance compared with RIFLE and AKIN. [1]
AKI is defined by any of the following:
-
Rise in serum creatinine ≥0.3 mg/dL within 48 hours
-
Rise in serum creatinine ≥1.5 times baseline, which is known or presumed to have occurred within the prior seven days
-
Urine output < 0.5 ml/kg/hour for six hours
The criteria for AKI stages are similar to AKIN, except for stage 3 AKI, which comprises an increase in serum creatinine of ≥0.3 mg/dL (rather than ≥ 0.5 mg/dL) to ≥4 mg/dL.
Cardiovascular complications
Cardiovascular complications (eg, heart failure, myocardial infarction, arrhythmias, cardiac arrest) have been observed in as many as 35% of patients with AKI. Fluid overload secondary to oliguric AKI is a particular risk for elderly patients with limited cardiac reserve. Additionally, AKI is associated with electrolyte and acid-base imbalance that can increase the risk of developing arrhythmia and decrease myocardial contractility. In cardiac patients who experience AKI either in the setting of acute decompensated heart failure or cardiac surgery, AKI is associated with worse morbidity and mortality. [5]
Pericarditis is a relatively rare complication of AKI. When pericarditis complicates AKI, consider additional diagnoses, such as systemic lupus erythematosus (SLE) and hepatorenal syndrome.
AKI also can be a complication of cardiac diseases, such as endocarditis, decompensated heart failure, or atrial fibrillation with emboli. Cardiac arrest in a patient with AKI should always arouse suspicion of hyperkalemia. Many authors recommend that in addition to ACLS measures in patients with PEA arrest, a trial of intravenous calcium chloride (or gluconate) should be considered in patients with AKI with known or suspected hyperkalemia.
Pulmonary complications
Pulmonary complications have been reported in approximately 54% of patients with AKI and are the single most significant risk factor for death in patients with AKI. Proposed mechanisms for acute lung injury during AKI include hypervolemia, increased proinflammatory cytokine levels, leukocyte infiltration, and increased pulmonary vascular permeability. In addition, diseases exist that commonly present with simultaneous pulmonary and renal involvement, including the following:
-
Goodpasture syndrome
-
Granulomatosis with polyangiitis (Wegener granulomatosis)
-
Polyarteritis nodosa
-
Cryoglobulinemia
-
Sarcoidosis
Hypoxia commonly occurs during hemodialysis and can be particularly significant in patients with pulmonary disease. This dialysis-related hypoxia is thought to occur secondary to white blood cell (WBC) lung sequestration and alveolar hypoventilation.
Gastrointestinal complications
Nausea, vomiting, and anorexia are frequent complications of AKI and represent one of the cardinal signs of uremia. GI bleeding occurs in approximately one-third of patients with AKI. Most episodes are mild, but GI bleeding accounts for 3-8% of deaths in patients with AKI.
Pancreatitis
Mild hyperamylasemia is commonly seen in AKI. Elevation of baseline amylase concentrations can complicate the diagnosis of pancreatitis in patients with AKI. Lipase measurement, frequently suppressed in AKI, should be considered in this light when there is suspicion of pancreatitis. Pancreatitis has been reported as a concurrent illness with AKI in patients with atheroemboli, vasculitis, and sepsis from ascending cholangitis.
Jaundice
Jaundice frequently complicates AKI. Etiologies of jaundice with AKI include hepatic congestion, blood transfusions, and sepsis.
Hepatitis
Hepatitis occurring concurrently with AKI should prompt consideration of the following disorders in the differential diagnosis:
-
Common bile duct obstruction
-
Fulminant hepatitis
-
HBV- and HCV-associated glomerulonephritis
-
Leptospirosis
-
Medication toxicity (eg, acetaminophen toxicity)
-
Amanita phalloides poisoning
Infectious complications
Infections commonly complicate the course of AKI and have been reported to occur in as many as 33% of patients with AKI. It is attributed to possible altered cytokine homeostasis and immune cell dysfunction associated with AKI. The most common sites of infection are the pulmonary and urinary tracts. Infections are the leading cause of morbidity and death in patients with AKI. Various studies have reported mortality rates of 11-72% in infections complicating AKI.
Neurologic complications
Neurologic symptoms of uremia have been reported in approximately 38% of patients with AKI. Neurologic sequelae include lethargy, somnolence, reversal of the sleep-wake cycle, and cognitive or memory deficits. Focal neurologic deficits are rarely caused solely by uremia.
The pathophysiology of neurologic symptoms is still unknown but is partially attributed to the possible accumulation of neurotoxic metabolites in patients with severe AKI that can lead to an imbalance in cellular water transportation and disturbance of the blood-brain barrier. However, these symptoms do not correlate well with levels of BUN or creatinine.
A number of diseases can present with concurrent neurologic and renal manifestations, including the following:
-
SLE
-
Thrombotic thrombocytopenic purpura (TTP)
-
Hemolytic uremic syndrome (HUS)
-
Endocarditis
-
Malignant hypertension
Also see Acute Kidney Injury (Renal Failure) in Emergency Medicine.
Pathophysiology
The driving force for glomerular filtration is the pressure gradient from the glomerulus to the Bowman space. Glomerular pressure depends primarily on renal blood flow (RBF) and is controlled by the combined resistances of renal afferent and efferent arterioles. Regardless of the cause of AKI, reductions in RBF represent a common pathologic pathway for a decrease in the glomerular filtration rate (GFR). The etiology of AKI consists of 3 main mechanisms: prerenal, intrinsic, and obstructive (postrenal).
In prerenal failure, GFR is depressed by compromised renal perfusion. Tubular and glomerular functions remain normal.
Intrinsic failure includes diseases of the kidney itself, predominantly affecting the glomerulus, interstitium, or tubule, which are associated with the release of renal afferent vasoconstrictors. Ischemia is the most common cause of intrinsic kidney failure. Patients with chronic kidney disease (CKD) may also present with superimposed AKI from prerenal failure and obstruction, as well as intrinsic kidney disease.
Obstruction of the urinary tract initially causes an increase in tubular pressure, which decreases the filtration driving force. This pressure gradient soon equalizes, and maintenance of a depressed GFR then depends on renal efferent vasoconstriction.
Depressed renal blood flow
Depressed RBF eventually leads to ischemia and cell death. This may happen without systemic hypotension is present and is referred to as normotensive ischemic AKI. The initial ischemic insult triggers a cascade of events, including production of oxygen free radicals, cytokines, and enzymes; endothelial activation and leukocyte adhesion; activation of coagulation; and initiation of apoptosis. These events continue to cause cell injury even after restoration of RBF.
Tubular cellular damage results in the disruption of tight junctions between cells, allowing back leak of glomerular filtrate and further depressing effective GFR. In addition, dying cells slough off into the tubules, forming obstructing casts, further decreasing GFR and leading to oliguria.
During this period of depressed RBF, the kidneys are particularly vulnerable to additional insults; this is when iatrogenic kidney injury is most common. The following are frequent combinations:
-
Radiocontrast agents, aminoglycosides, or cardiovascular surgery with preexisting kidney disease (eg, elderly, diabetic, jaundiced patients)
-
Angiotensin-converting enzyme (ACE) inhibitors with diuretics, small- or large-vessel renal arterial disease
-
Nonsteroidal anti-inflammatory drugs (NSAIDs) with chronic heart failure, hypertension, or renal artery stenosis
Acute tubular necrosis
Frank necrosis is not prominent in most cases of acute tubular necrosis (ATN) and tends to be patchy. The following pathologic changes can be seen following ATN injury (see the image below):
-
Loss of brush border
-
Flattening of the epithelium
-
Detachment of cells
-
Presence of intratubular casts
-
Dilatation of the lumen
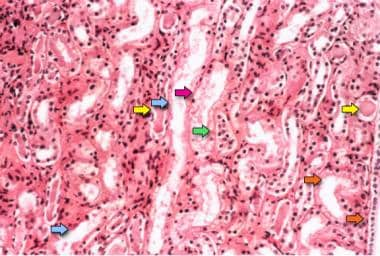 Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
Although these changes are observed predominantly in proximal tubules, injury to the distal nephron can also be demonstrated. In addition, the distal nephron may become obstructed by desquamated cells and cellular debris. See the image above.
Apoptosis
In contrast to necrosis, the distal nephron is the principal site of apoptotic cell death. During the initial phase of ischemic injury, loss of integrity of the actin cytoskeleton leads to flattening of the epithelium, with loss of the brush border, loss of focal cell contacts, and subsequent disengagement of the cell from the underlying substratum.
Inflammatory response
Many endogenous growth factors that participate in the regeneration process following ischemic renal injury have not been identified. However, the administration of growth factors exogenously has been shown to ameliorate and hasten recovery from AKI.
Depletion of neutrophils and blockage of neutrophil adhesion reduces renal injury following ischemia, indicating that the inflammatory response is responsible, in part, for some features of ATN, especially in postischemic injury after transplant.
Vasoconstriction
Intrarenal vasoconstriction is the dominant mechanism for reduced GFR in patients with ATN. The mediators of this vasoconstriction are unknown, but tubular injury seems to be an important concomitant finding. Urine backflow and intratubular obstruction (from sloughed cells and debris) are causes of reduced net ultrafiltration. The importance of this mechanism is highlighted by the improvement in renal function that follows the relief of such intratubular obstruction.
In addition, when obstruction is prolonged, intrarenal vasoconstriction is prominent in part due to the tubuloglomerular feedback mechanism, which is thought to be mediated by adenosine and activated when there is proximal tubular damage. The macula densa senses the increased chloride load and feeds back to cause arteriolar vasoconstriction.
Apart from the increased basal renal vascular tone, the stressed renal microvasculature is more sensitive to potentially vasoconstrictive drugs and otherwise-tolerated changes in systemic blood pressure. The vasculature of the injured kidney has an impaired vasodilatory response and loses its autoregulatory behavior.
This latter phenomenon has important clinical relevance because the frequent reduction in systemic pressure during intermittent hemodialysis may provoke additional damage that can delay recovery from ATN. Often, injury results in atubular glomeruli, where the glomerular function is preserved, but the lack of tubular outflow precludes its function.
Isosthenuria
A physiologic hallmark of ATN is a failure to dilute or concentrate urine (isosthenuria) maximally. This defect is not responsive to pharmacologic doses of vasopressin. The injured kidney fails to generate and maintain a high medullary solute gradient because solute accumulation in the medulla depends on normal distal nephron function.
Failure to excrete concentrated urine, even in the presence of oliguria, is a helpful diagnostic clue in distinguishing prerenal from intrinsic AKI. In prerenal azotemia, urine osmolality is typically more than 500 mOsm/kg, whereas, in intrinsic kidney disease, urine osmolality is less than 300 mOsm/kg.
Restoration of renal blood flow and associated complications
Recovery from AKI is first dependent upon the restoration of RBF. Early RBF normalization predicts a better prognosis for recovery of renal function. In prerenal failure, restoration of circulating blood volume is usually sufficient. Rapid relief of urinary obstruction in postrenal failure results in a prompt decrease of vasoconstriction. With intrinsic renal failure, removing tubular toxins and initiating therapy for glomerular diseases decreases renal afferent vasoconstriction.
Once RBF is restored, the remaining functional nephrons increase their filtration and eventually undergo hypertrophy. GFR recovery depends on the size of this remnant nephron pool. If the number of remaining nephrons is below a critical threshold, continued hyperfiltration results in progressive glomerular sclerosis, eventually leading to increased nephron loss.
A vicious cycle ensues: continued nephron loss causes more hyperfiltration until complete kidney failure results. This has been termed the hyperfiltration theory of kidney failure and explains the scenario in which progressive failure is frequently observed after apparent recovery from AKI.
Etiology
Prerenal AKI
Prerenal AKI represents the most common form of kidney injury and often leads to intrinsic AKI if it is not promptly corrected. Volume loss can provoke this syndrome; the source of the loss may be GI, renal, or cutaneous (eg, burns) or from internal or external hemorrhage. Prerenal AKI can also result from decreased renal perfusion in patients with heart failure or shock (eg, sepsis, anaphylaxis). In patients taking calcium channel blockers, use of the antibiotic clarithromycin can result in AKI, due to a drug-drug interaction that markedly raises plasma calcium channel blocker concentrations and causes hypotension, with subsequent ischemic damage to the kidney. [6]
Several classes of medications can induce prerenal AKI in volume-depleted states, including ACE inhibitors and angiotensin receptor blockers (ARBs), which are otherwise safely tolerated and beneficial in most patients with chronic kidney disease (CKD). Aminoglycosides, amphotericin B, and radiologic contrast agents may also do so.
Arteriolar vasoconstriction leading to prerenal AKI can occur in hypercalcemic states, as well as with the use of radiocontrast agents, NSAIDs, amphotericin, calcineurin inhibitors, norepinephrine, and other pressor agents. The hepatorenal syndrome can also be considered a form of prerenal AKI, because functional kidney failure develops from diffuse vasoconstriction in vessels supplying the kidney. [7]
To summarize, volume depletion can be caused by the following:
-
Renal losses - Diuretics, polyuria
-
GI losses - Vomiting, diarrhea
-
Cutaneous losses - Burns, Stevens-Johnson syndrome
-
Hemorrhage
-
Pancreatitis
Decreased cardiac output can be caused by the following:
-
Heart failure
-
Pericardial effusion/tamponade
-
Pulmonary embolus
-
Acute myocardial infarction
-
Severe valvular disease
-
Abdominal compartment syndrome - Tense ascites
Systemic vasodilation can be caused by the following:
-
Sepsis
-
Anaphylaxis
-
Anesthetics
-
Drug overdose
-
Cancer-specific etiologies: Capillary leak syndrome, engraftment syndrome, chimeric antigen receptor (CAR) T-cell therapy, all-trans retinoic acid (ATRA), gemcitabine, interleukin-2 (IL-2), granulocyte-macrophage colony-stimulating factor (GM-CSF)
Afferent arteriolar vasoconstriction can be caused by the following:
-
Hypercalcemia
-
Drugs - NSAIDs, amphotericin B, calcineurin inhibitors, norepinephrine, radiocontrast agents
-
Hepatorenal syndrome and and sinusoidal obstruction syndrome (post hematopoietic stem cell transplantation [HSCT])
Diseases that decrease effective arterial blood volume include the following:
-
Hypovolemia
-
Heart failure
-
Liver failure
-
Sepsis
Renal arterial diseases that can result in AKI include renal arterial stenosis, especially in the setting of hypotension or initiation of ACE inhibitors or ARBs. Renal artery stenosis typically results from atherosclerosis or fibromuscular dysplasia, but is also a feature of the genetic syndromes type 1 neurofibromatosis, Williams syndrome, and Alagille syndrome.
Patients can also develop septic embolic disease (eg, from endocarditis) or cholesterol emboli, often as a result of instrumentation or cardiovascular surgery.
Intrinsic AKI
Structural injury in the kidney is the hallmark of intrinsic AKI; the most common form is ATN, either ischemic or cytotoxic. Glomerulonephritis can be a cause of AKI and usually falls into a class referred to as rapidly progressive glomerulonephritis (RPGN). Glomerular crescents (glomerular injury) are found in RPGN on biopsy; presence of crescents in more than 50% of glomeruli usually corresponds to a significant decline in renal function. Although comparatively rare, acute glomerulonephritides should be part of the diagnostic consideration in cases of AKI.
To summarize, vascular (large- and small-vessel) causes of intrinsic AKI include the following:
-
Renal artery obstruction - Thrombosis, emboli, dissection, vasculitis
-
Renal vein obstruction - Thrombosis
-
Microangiopathy - TTP, HUS, disseminated intravascular coagulation (DIC), preeclampsia
-
Malignant hypertension
-
Scleroderma renal crisis
-
Transplant rejection
-
Atheroembolic disease
Glomerular causes include the following:
-
Anti–glomerular basement membrane (GBM) disease - As part of Goodpasture syndrome or renal limited disease
-
Pauci-immune glomerulonephritis - Antineutrophil cytoplasmic antibody (ANCA)–associated glomerulonephritis such granulomatosis with polyangiitis (Wegener granulomatosis), eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), microscopic polyangiitis
-
Immune complex glomerulonephritis - Lupus nephritis, postinfectious glomerulonephritis, IgA nephropathy, cryoglobulinemia, primary membranoproliferative glomerulonephritis
Tubular etiologies may include ischemia or cytotoxicity. Cytotoxic etiologies include the following:
-
Heme pigment - Rhabdomyolysis, intravascular hemolysis
-
Crystals - Tumor lysis syndrome, dysproteinemia, seizures, ethylene glycol poisoning, megadose vitamin C, acyclovir, indinavir, methotrexate
-
Drugs - Aminoglycosides, lithium, amphotericin B, pentamidine, cisplatin, pemetrexed, zoledronic acid, ifosfamide, radiocontrast agents
-
Lysozyme-induced nephropathy
Interstitial causes include the following:
-
Drugs - Penicillins, cephalosporins, NSAIDs, proton pump inhibitors, allopurinol, rifampin, indinavir, mesalamine, sulfonamides [8]
-
Infection - Pyelonephritis, viral nephritides (eg, BK virus nephropathy in HSCT recipients)
-
Immunotherapy and targeted therapy - Immune checkpoint inhibitors (ICIs), lenalidomide, certain tyrosine kinase inhibitors (TKIs), BRAF inhibitors
-
Systemic disease - Sjögren syndrome, sarcoid, lupus, lymphoma, leukemia, tubulonephritis, uveitis
Anticoagulant-related nephropathy is a form of AKI in which over-anticoagulation causes profuse glomerular hemorrhage. Kidney biopsies in these patients show red blood cells and red cell casts filling numerous renal tubules. [9] Studies of anticoagulation for atrial fibrillation have shown that in elderly and Asian patients, the risk of anticoagulant-related nephropathy is greater with warfarin than with direct oral anticoagulants (eg, apixaban, rivaroxaban, dabigatran). [10, 11]
Postrenal AKI
Mechanical obstruction of the urinary collecting system, including the renal pelvis, ureters, bladder, or urethra, results in obstructive uropathy or postrenal AKI. Causes of obstruction include the following:
-
Stone disease
-
Stricture
-
Intraluminal, extraluminal, or intramural tumors
-
Thrombosis or compressive hematoma
-
Fibrosis
If the site of obstruction is unilateral, then a rise in the serum creatinine level may not be apparent because of the preserved function of the contralateral kidney. Nevertheless, even with unilateral obstruction, a significant loss of GFR occurs, and patients with partial obstruction may develop progressive loss of GFR if the obstruction is not relieved.
Bilateral obstruction is usually a result of prostate enlargement or tumors in men and urologic or gynecologic tumors in women. Patients who develop anuria typically have an obstruction at the level of the bladder or downstream to it.
To summarize, causes of postrenal AKI include the following:
-
Ureteric obstruction - Stone disease, tumor, fibrosis, ligation during pelvic surgery
-
Bladder neck obstruction - Benign prostatic hyperplasia (BPH), prostate cancer, neurogenic bladder, tricyclic antidepressants, ganglion blockers, bladder tumor, stone disease, hemorrhage/clot
-
Urethral obstruction - Strictures, tumor, phimosis
-
Intra-abdominal hypertension - Tense ascites
-
Renal vein thrombosis
Diseases causing urinary obstruction from the level of the renal tubules to the urethra include the following:
-
Tubular obstruction from crystals - Eg, uric acid, calcium oxalate, acyclovir, sulfonamide, methotrexate, myeloma light chains
-
Ureteral obstruction - Retroperitoneal tumor, retroperitoneal fibrosis (methysergide, propranolol, hydralazine), urolithiasis, papillary necrosis, BK virus infection
-
Urethral obstruction - BPH; prostate, cervical, bladder, or colorectal carcinoma; bladder hematoma; bladder stone; obstructed Foley catheter; neurogenic bladder; stricture
Etiology in newborns and infants
Prerenal AKI
The patient's age has significant implications for the differential diagnosis of AKI. In newborns and infants, causes of prerenal AKI include the following:
-
Perinatal hemorrhage - Twin-twin transfusion, complications of amniocentesis, abruptio placenta, birth trauma
-
Neonatal hemorrhage - Severe intraventricular hemorrhage, adrenal hemorrhage
-
Perinatal asphyxia and hyaline membrane disease (newborn respiratory distress syndrome) - Both may result in preferential blood shunting away from the kidneys to the central circulation
Intrinsic AKI
Causes of intrinsic AKI include the following:
-
ATN - Can occur in the setting of perinatal asphyxia; ATN also has been observed secondary to medications (eg, aminoglycosides, NSAIDs) given to the mother perinatally
-
ACE inhibitors - Can traverse the placenta, resulting in a hemodynamically mediated form of AKI
-
Acute glomerulonephritis – Rare; most commonly the result of maternal-fetal transfer of antibodies against the neonate's glomeruli or transmission of chronic infections (syphilis, cytomegalovirus) associated with acute glomerulonephritis
Postrenal AKI
Congenital malformations of the urinary collecting systems should be suspected in cases of postrenal AKI.
Etiology in children
Prerenal AKI
In children, gastroenteritis is the most common cause of hypovolemia and can result in prerenal AKI. Congenital and acquired heart diseases are also important causes of decreased renal perfusion in this age group.
Intrinsic AKI
Intrinsic AKI may result from any of the following:
-
Acute poststreptococcal glomerulonephritis - Should be considered in any child who presents with hypertension, edema, hematuria, and kidney failure
-
HUS - Often cited as the most common cause of AKI in children
The most common form of HUS is associated with a diarrheal prodrome caused by Escherichia coli O157:H7. These children usually present with microangiopathic anemia, thrombocytopenia, colitis, mental status changes, and kidney failure. TTP is not as strongly associated with AKI.
In a study of 521 pediatric trauma patients with posttraumatic rhabdomyolysis, AKI occurred in 70 (13.4%) patients. Independent risk factors for AKI were a creatine kinase level of ≥3,000, an Injury Severity Score of ≤15, a Glasgow Coma Scale score of ≤8, an abdominal Abbreviated Injury Scale (AIS) score of ≤3, imaging studies with contrast, blunt mechanism of injury, administration of nephrotoxic agents, and requirement for the administration of fluids in the emergency department. [12]
Cardiopulmonary bypass and AKI
Longer time on extracorporeal cardiopulmonary bypass is commonly accepted as a risk factor for AKI. However, a study by Mancini et al. found that extracorporeal cardiopulmonary bypass time did not predict AKI requiring dialysis, suggesting that a risk assessment may be a more reliable marker. [13] Achieving moderate glucose control and using balanced crystalloid solutions perioperatively have been associated with decreased risk of AKI. [14, 15] Similarly, implementing the KDIGO “bundle of care” in high-risk patients has been associated with decreased risk of AKI postoperatively. [16] This bundle consists of the following:
-
Discontinuation of nephrotoxic agents for 48 hours postoperatively
-
Avoidance of hyperglycemia for 72 hours postoperatively
-
Close monitoring of UOP and serum creatinine
-
Consideration of alternatives to radiocontrast agent
-
Optimization of volume status and perfusion pressure using a prespecified algorithm
-
Functional hemodynamic monitoring
COVID-19 and AKI
Kidney involvement is frequent in patients with severe COVID-19. More than 40% of patients have proteinuria on hospital admission, and approximately 20–40% of patients admitted to intensive care units in Europe and the United States have AKI. [17] In patients with COVID-19, severe AKI is an ominous development associated with high mortality. In a study of over 89,000 US veterans who were 30-day survivors of COVID-19, the risk of AKI, estimated GFR decline, and ESKD were significantly greater than non-infected controls, and those who were hospitalized and admitted to the intensive care unit had the highest risk for adverse renal outcomes. [18, 19]
AKI in COVID-19 is multifactorial and may have a distinct pathophysiology that includes the following [20, 21] :
-
Hypotension and decreased kidney perfusion secondary to hemodynamic or hemostatic factors or associated sepsis
-
Endothelial dysfunction, with hypercoagulability and complement activation
-
Nephrotoxins (medications, contrast agents)
-
Organ crosstalk among the injured lungs, heart, and kidney
-
Direct infection of kidney tubules with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that induces cytoplasmic renal tubular inclusions; this can explain both AKI in acute COVID-19 and the development of CKD in individuals with long COVID. [22]
Cancer and AKI
Cancer patients have an increased risk of developing AKI due to multiple risk factors, including old age, chemotherapy/immunotherapy-associated nephrotoxicity, increased prevalence of CKD, and factors associated with cancer itself. [23] The reported overall risk of developing AKI during hospitalization for cancer has ranged from 12 to 21%, with the majority of cases, 50-75%, being mild (stage 1) and with less than 5% of the patients requiring renal replacement therapy (RRT). [23, 24, 25] However, the incidence of AKI can increase up to 68% in patients with hematologic malignancy and in a critical care setting. Patients with hematologic, renal, hepatic, and gastrointestinal malignancy have the highest rate of AKI. [24, 25] Hypovolemia and ATN are the most common etiologies, as they are in non-cancer patients, with sepsis and nephrotoxins being the leading causes of ATN. [26, 27] Other cancer-specific etiologoies are listed below. [28, 29, 30, 31]
Prerenal AKI related to cancer may have the following causes:
-
Cardio-renal: Malignant pericardial effusion, cardiotoxic chemotherapy (eg, trastuzumab, anthracyclines such as doxorubicin)
-
Hepato-renal: Budd Chiari syndrome, extensive metastatic liver disease, hepatic veno-occlusive disease in HSCT recipients
-
Systemic: Cytokine release syndrome/capillary leak syndrome (engraftment syndrome post HSCT), certain cancer therapies (eg, CAR T-cell, ATRA, gemcitabine, IL-2)
Intrinsic AKI related to cancer may have the following causes:
-
Glomerular pathology: Paraneoplastic glomerulonephritis or medication-associated glomerulonephritis (eg, from anthracyclines, pamidronate, interferon, mechanistic target of rapamycin [mTOR] inhibitors, TKIs, ICIs)
-
Thrombotic microangiopathy (TMA): Paraneoplastic TMA, medication-associated TMA (eg, from carfilzomib, bortezomib, cisplatin, gemcitabine, anthracycline, VGEF-targeting agents, TKIs, mTOR inhibitors)
-
Tubulointerstitial injury: Tumor infiltration or metastasis, lenalidomide, BRAF inhibitors, BK virus nephropathy in HSCT recipients, TKIs, ICIs
-
Acute tubular injury: Lysozyme-induced nephropathy, cytokine release syndrome/capillary leak syndrome, tumor lysis syndrome, medications (eg, cisplatin, pemetrexed, ifosfamide, CAR T-cell therapy)
-
Crystal nephropathy: Tumor lysis syndrome, multiple myeloma, methotrexate
Postrenal AKI related to cancer may have the following causes:
-
Ureteral compression (by obstructing tumor, lymphadenopathy, retroperitoneal fibrosis, or BK virus infection), blood clots
-
Urinary bladder obstruction
Recovery from AKI is variable and depends on the underlying etiology. In the majority of patients, AKI resolves after hospital discharge. Up to 23% of the patients with severe AKI requiring RRT who survive critical illness are expected to require long-term hemodialysis. [25, 32, 33]
In addition, AKI is associated with increased mortality (up to 6-fold), increased hospital length of stay, cost of treatment, hematologic malignancy relapse, and higher mortality rates than in cancer patients with no AKI. [23, 26, 32, 34, 35]
Epidemiology
In the United States, approximately 1% of patients admitted to hospitals have AKI at the time of admission. The estimated incidence rate of AKI during hospitalization is 2-5%. AKI develops within 30 days postoperatively in approximately 1% of general surgery cases and arises in more than 50% of intensive care unit (ICU) patients. [36, 37] In recipients of solitary kidney transplants, 21% developed AKI within the first 6 months after transplantation. [38]
Harding et al calculated that in the United States from 2000 to 2015, hospitalization rates for dialysis-requiring AKI in adults increased considerably while mortality decreased. In adults with diabetes, rates increased from 26.4 to 41.1 per 100,000 population, with relative increases greater in younger versus older adults. In adults without diabetes, rates increased from 4.8 to 8.7 per 100,000 population between 2000 and 2009, and then plateaued. Mortality declined significantly in patients both with and without diabetes. [39]
In a prospective national cohort study in Wales that used an electronic AKI alert (a centralized laboratory system that automatically compares measured creatinine values in an individual patient with previous results to generate alerts), the incidence of AKI was 577 per 100,000 population. Community-acquired AKI accounted for 49.3% of all incident episodes, and 42% occurred in the context of preexisting chronic kidney disease. The 90-day mortality rate was 25.6%, and 23.7% of episodes progressed to a higher AKI stage. [40]
In a Canadian study of severely ill children admitted to pediatric intensive care units, 30.3% developed AKI and 12.2% developed severe AKI. The incidence rate for critical illness–associated AKI was 34 per 100,000 children-year, and the rate of severe AKI was 14 per 100,000 children-year. Severe AKI was more common in boys (incidence rate ratio, 1.55) and in infants younger than 1 year old (incidence rate ratio, 14.77). The AKI-associated mortality rate was 2.3 per 100,000 children-year. [41]
Approximately 95% of consultations with nephrologists are related to AKI. Feest and colleagues calculated that the appropriate nephrologist referral rate is approximately 70 cases per million population. [42]
Prognosis
The prognosis for patients with AKI is directly related to the cause of the injury and, to a great extent, to the presence or absence of preexisting kidney disease (estimated GFR [eGFR] < 60 mL/min), as well as to the duration of kidney dysfunction prior to therapeutic intervention. In the past, AKI was thought to be completely reversible, but long-term follow-up of patients with this condition has shown otherwise.
A study from Canada showed a much higher incidence of AKI than did previous reports, with a rate of 18.3% (7856 of 43,008) in hospitalized patients. [43] The incidence of AKI correlated inversely with eGFR and was associated with a higher mortality rate and a higher incidence of subsequent end-stage kidney disease (ESKD) at each level of baseline eGFR.
However, the greatest impact on mortality was seen in individuals with an eGFR of greater than 60 mL/min who developed AKI. Those with stage 3 AKI (AKIN criteria; see Background) had a mortality rate of 50%, while mortality in individuals with an eGFR of greater than 60 mL/min but who did not develop AKI was only 3%. Among individuals with an eGFR of less than 30, the mortality rate was 12.1% in those who did not develop AKI, versus 40.7% among patients with stage 3 AKI. [43]
In one study, survivors of severe AKI had worse health-related quality of life (QOL) than the general population, even after adjusting for their reduced kidney function. Both physical and mental components were affected. Increasing age and reduced kidney function were associated with poorer physical QOL. [44]
Mortality rates and associated factors
If AKI is defined by a sudden increment of serum creatinine of 0.5-1 mg/dL and is associated with a mild to moderate rise in creatinine, the prognosis tends to be worse. (Increments of 0.3 mg/dL in serum creatinine, especially at lower ranges of serum creatinine, have important prognostic significance).
The mortality rate for ICU patients with AKI is higher (> 50% in most studies), particularly when AKI is severe enough to require dialysis treatment. [45] ICU patients with sepsis-associated AKI have significantly higher mortality rates than do nonseptic AKI patients. [46]
In addition, the pooled estimate for general ICU patients with AKI shows a stepwise increase in relative risk for death through the risk, injury, and failure classifications of the RIFLE criteria in AKI patients versus non-AKI patients. [47] This reflects the fact that the high mortality rate in patients with AKI who require dialysis may not be related to the dialysis procedure or accompanying comorbidities and that AKI is an independent indicator of mortality. The survival rate is nearly 0% among patients with AKI who have an Acute Physiology and Chronic Health Evaluation II (APACHE II) score higher than 40. In patients with APACHE II scores of 10-19, the survival rate is 40%.
Fluid balance and mortality
In a post hoc analysis of the Fluid and Catheter Treatment Trial (FACTT), which examined liberal versus conservative fluid management in intubated ICU patients, fluid balance and diuretic use were identified as prognostic factors for mortality in individuals with AKI. Specifically, greater cumulative fluid accumulation over an average of 6 days (10.2 L vs 3.7 L in the liberal vs conservative group, respectively) was associated with a higher mortality rate, and higher furosemide use (cumulatively, 562 mg vs 159 mg, respectively) was associated with a lower mortality rate. [48]
Of note, more than half of the individuals in FACTT had stage 1 AKI (AKIN criteria), so whether these results apply to more severe AKI stages is unclear. One interpretation of this study is that patients who can be stabilized with less volume resuscitation fare better. From a practical standpoint, one conclusion is that aggressive, prolonged volume resuscitation does not improve prognosis in AKI in the ICU setting. [48]
Additional prognostic factors
Other prognostic factors include the following:
-
Older age
-
Multiorgan failure - Ie, the more organs that fail, the worse the prognosis
-
Oliguria
-
Hypotension
-
Vasopressor support
-
Number of transfusions
-
Noncavitary surgery
-
Occurrence of AKI by itself [49] - Has significant negative prognostic implications
Prerenal azotemia from volume contraction is treated with volume expansion; if left untreated for a prolonged period, tubular necrosis may result and may not be reversible. If left untreated for a long time, postrenal AKI may result in irreversible kidney damage. Procedures such as catheter placement, lithotripsy, prostatectomy, stent placement, and percutaneous nephrostomy can help to prevent permanent kidney damage.
Nephritis
Timely identification of pyelonephritis, proper treatment, and further prevention using prophylactic antibiotics may improve the prognosis, especially in females. Early diagnosis of acute interstitial nephritis and crescentic glomerulonephritis via kidney biopsy and other appropriate tests may enhance early renal recovery because appropriate therapy can be initiated promptly and aggressively. For example, the number of crescents, the type of crescents (ie, cellular vs fibrous), and the serum creatinine level at the time of presentation may dictate the prognosis for renal recovery in these patients.
Proteinuria
A large cohort study demonstrated that proteinuria coupled with low baseline GFR is associated with a higher incidence of AKI and should be considered as an identifying factor for individuals at risk. [49] A retrospective, population-based study in a cohort of patients with and without known preoperative kidney dysfunction undergoing elective inpatient surgery found that proteinuria was associated with postoperative AKI and 30-day unplanned readmission independent of preoperative eGFR. [50]
Statins
The relationship between statins and AKI is complex. [51] In addition to rare cases of statins causing rhabdomyolysis, the use of high-potency statins has been associated with an increased rate of diagnosis for AKI in hospital admissions, compared with the use of low-potency statins, particularly in the first 120 days after initiation of statin treatment. [52] On the other hand, preprocedural statin therapy has been shown to reduce contrast-induced AKI in patients undergoing coronary angiography. [53, 54]
Research on perioperative statins has yielded mixed results. A retrospective study in more than 200,000 patients older than 66 years who underwent elective surgery suggested that patients taking statins had a lesser incidence and lower severity of AKI, as well as lower mortality, than did individuals not on statins. [55] In a meta-analysis of patients undergoing major surgery, preoperative statin therapy was associated with a significant risk reduction for cumulative postoperative AKI and postoperative AKI requiring renal replacement therapy. Still, when the analysis was restricted to randomized controlled trials, the protective effect was not significant. [56]
A meta-analysis in adult patients who required surgery with cardiac bypass found no association between preoperative statin use and a decrease in the incidence of AKI. [57] Similarly, a meta-analysis in patients undergoing cardiac surgery (mainly myocardial revascularization) found that preoperative statin treatment did not influence perioperative kidney failure. [58] In contrast, in another meta-analysis of patients undergoing cardiac surgery, preoperative statin therapy significantly reduced the incidence of postoperative kidney dysfunction and the need for postoperative renal replacement therapy. [59]
Long-term prognosis
In contrast to previous beliefs, it is now known that survivors of AKI do not universally have a benign course. On long-term follow-up (1-10 years), approximately 12.5% of survivors of AKI are dialysis dependent; rates range widely, from 1-64%, depending on the patient population. From 19-31% of survivors experience partial recovery of kidney function and have chronic kidney disease. [37]
In a long-term follow-up study of 350 patients from the randomized RENAL trial who survived AKI in the ICU, researchers found that the overall mortality rate was 62% at a median of 42.4 months after randomization. Median survival did not significantly differ between patients who received high- or low-intensity renal replacement therapy. At follow-up, 42.1% of the surviving patients had microalbuminuria or macroalbuminuria. Only 5.4% of the patients surviving at day 90 required maintenance dialysis. Predictors of long-term mortality included age, APACHE III score, and serum creatinine levels at baseline. [60] In patients who survived AKI, cancer and cardiovascular disease is the most common etiology for death after hospitalization. [61]
Patient Education
Educating patients about the nephrotoxic potential of common therapeutic agents is always helpful. Nonsteroidal anti-inflammatory drugs (NSAIDs) provide a good example; most patients are unaware of their nephrotoxicity, and their universal availability makes them a constant concern.
For patient education information, see Acute Kidney Failure.
-
Pigmented, muddy brown, granular casts are visible in the urine sediment of a patient with acute tubular necrosis (400x magnification).
-
Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
Tables
Stage |
GFR Criteria |
Urine Output Criteria |
Probability |
Risk |
SCreat increased × 1.5 or GFR decreased >25% |
UO < 0.5 mL/kg/h × 6 h |
High sensitivity (Risk >Injury >Failure) |
Injury |
SCreat increased × 2 or GFR decreased >50% |
UO < 0.5 mL/kg/h × 12 h |
|
Failure |
SCreat increased × 3 or GFR decreased 75% or SCreat ≥4 mg/dL; acute rise ≥0.5 mg/dL |
UO < 0.3 mL/kg/h × 24 h (oliguria) or anuria × 12 h |
|
Loss |
Persistent acute renal failure: complete loss of kidney function >4 wk |
High specificity |
|
ESKD |
Complete loss of kidney function >3 mo |
||
ESKD—end-stage kidney disease; GFR—glomerular filtration rate; SCreat—serum creatinine; UO—urine output Note: Patients can be classified by GFR criteria and/or UO criteria. The criteria that support the most severe classification should be used. The superimposition of acute on chronic failure is indicated with the designation RIFLE-FC; failure is present in such cases even if the increase in SCreat is less than 3-fold, provided that the new SCreat is greater than 4.0 mg/dL (350 µmol/L) and results from an acute increase of at least 0.5 mg/dL (44 µmol/L). |
|||
Stage |
Serum Creatinine Criteria |
Urine Output Criteria |
1 |
Increase of ≥0.3 mg/dL (≥26.4 µmol/L) or 1.5- to 2-fold increase from baseline |
< 0.5 mL/kg/h for >6 h |
2 |
> 2-fold to 3-fold increase from baseline |
< 0.5 mL/kg/h for >12 h |
3* |
> 3-fold increase from baseline, or increase of ≥ 4.0 mg/dL (≥35.4 µmol/L) with an acute increase of at least 0.5 mg/dL (44 µmol/L) |
< 0.3 mL/kg/h for 24 h or anuria for 12 h |
*Patients who receive renal replacement therapy (RRT) are considered to have met the criteria for stage 3 irrespective of the stage they are in at the time of RRT. |
||