Practice Essentials
Familial hypercholesterolemia (FH) is an autosomal dominant disorder that causes severe elevations in total cholesterol and low-density lipoprotein cholesterol (LDLc). [1, 2, 3]
Xanthomas are noted commonly on the Achilles tendons and metacarpal phalangeal extensor tendons of the hands of patients with untreated FH. See the image below.
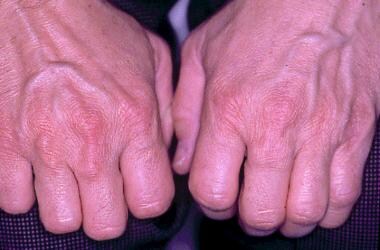 Metacarpophalangeal joint tendon xanthomas in a 45-year-old man with heterozygous familial hypercholesterolemia.
Metacarpophalangeal joint tendon xanthomas in a 45-year-old man with heterozygous familial hypercholesterolemia.
Signs and symptoms of familial hypercholesterolemia
Homozygous FH
Signs and symptoms of homozygous FH in children include the following:
-
Symptoms consistent with ischemic heart disease, peripheral vascular disease, cerebrovascular disease, or aortic stenosis
-
Articular symptoms such as tendonitis or arthralgias
-
Unusual skin lesions, such as cutaneous xanthomas at birth or by early childhood (eg, planar xanthomas, tuberous xanthomas; later, tendon xanthomas)
-
Corneal arcus may be present and is sometimes circumferential
-
Murmur of aortic stenosis may be present
Most patients with homozygous FH do not survive adulthood beyond age 30 years unless treated with unusual methods, such as liver transplantation, LDL apheresis, or ileal bypass surgery to dramatically lower their LDLc levels.
Heterozygous FH
Children with heterozygous FH do not have symptoms related to coronary heart disease (CHD), and most do not develop tendon xanthomas or corneal arcus. However, one parent will have severe hypercholesterolemia and will also probably have either a personal or family history for premature coronary artery disease (CAD).
Signs and symptoms of heterozygous FH in adults include the following:
-
Long-standing history of severe hypercholesterolemia dating back to childhood
-
If no previous acute coronary event, symptoms consistent with ischemic heart disease, especially in the presence of other cardiovascular risk factors (especially smoking)
-
Past or present symptoms of recurrent Achilles tendonitis or arthritic complaints
-
If heterozygous FH is left untreated, tendon xanthomas (Achilles tendons, metacarpophalangeal [MCP] extensor tendons) will occur by the third decade of life in more than 60% of patients
-
Xanthelasmas
See Clinical Presentation for more details.
Diagnosis of familial hypercholesterolemia
The diagnosis of both homozygous and heterozygous FH is based primarily on the finding of severe LDLc elevations in the absence of secondary causes of hypercholesterolemia.
A probable diagnosis of heterozygous FH can be made if the LDLc level is greater than 330 mg/dL or if tendon xanthomas are present in a patient with an LDLc level above the 95th percentile. Definitive diagnosis can be made only with gene or receptor analysis. However, a substantial increase in serum triglyceride levels should raise the possibility of another lipid disorder.
Testing
Findings on lipid analysis in patients with FH include the following:
-
Homozygous FH: Severely elevated cholesterol levels (total cholesterol and LDLc levels >600 mg/dL); triglyceride levels within the reference range
-
Heterozygous FH: Elevated LDLc levels commonly greater than 250 mg/dL; in patients younger than 20 years, an LDLc level higher than 200 mg/dL is highly suggestive of heterozygous FH or, possibly, familial ligand defective apoB-100; in adults, LDLc levels higher than 290-300 mg/dL suggest heterozygous FH
LDL receptor analysis can be used to identify the specific LDL receptor defect, and LDL receptor or apoB-100 studies can help distinguish heterozygous FH from the similar syndrome of familial defective apoB-100.
In August 2013, the European Atherosclerosis Society (EAS) published a consensus statement for screening and treatment of heterozygous FH. [4, 5] The recommendations for screening for heterozygous FH include patients with [4, 5] :
-
A family member presenting with diagnosed FH;
-
Plasma cholesterol in an adult ≥8mmol/L (≥310 mg/dL);
-
Plasma cholesterol in a child ≥6mmol/L (≥230 mg/dL);
-
Premature CHD;
-
Tendon xanthomas; or
-
Sudden premature cardiac death.
See Workup for more detail.
Management
The goal of FH treatment is to reduce the risk of CHD or risk of a CHD-equivalent condition (eg, carotid artery disease, diabetes, peripheral arterial disease). [6, 7, 8]
Risk categories for developing CHD are as follows:
-
High risk: CHD or CHD risk equivalent (10-year risk >20%)
-
Moderately high risk: More than 2 risk factors (10-year risk 10-20%)
-
Moderate risk: More than 2 risk factors (10-year risk 10%)
-
Lower risk: 0-1 risk factor
Homozygous FH
The following are used in the management of homozygous FH:
-
High doses of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) combined with bile acid sequestrants, ezetimibe, and niacin [11]
-
Anti–proprotein convertase subtilisin/kexin type 9 (anti-PCSK9) monoclonal antibodies (eg, evolocumab and alirocumab) can be used as an adjunct to diet and other LDLc-lowering therapies, [12] or
-
Evinacumab, or
-
Lomitapide in severe cases (with or without LDL apheresis)
-
LDL apheresis for selective removal of lipoproteins that contain apo-B (when the LDL receptors are absent or nonfunctional)
-
Estrogen replacement therapy in postmenopausal women
The following are procedures used in the treatment of homozygous FH:
-
Portacaval anastomosis
-
Liver transplantation (rarely)
Investigative therapies for homozygous and heterozygous FH include probucol, which causes regression of cutaneous and tendon xanthomas in patients with both homozygous and heterozygous FH but no long-term benefits for reduced coronary atherosclerosis, and gene therapy.
Heterozygous FH
The following are used in the management of heterozygous FH:
-
Lifestyle modification, including diet (limited saturated fats, trans fats, and cholesterol); weight management; aerobic/toning exercises
-
HMG-CoA reductase inhibitors (statins) (eg, simvastatin, atorvastatin, or rosuvastatin), and one or more other LDL lowering medications, or
-
Anti–proprotein convertase subtilisin/kexin type 9 (anti-PCSK9) monoclonal antibodies (eg, evolocumab and alirocumab) can be used as an adjunct to diet and other LDLc-lowering therapies, or
-
Adenosine triphosphate-citrate lyase (ACL) inhibitor (eg, bempedoic acid) added to maximally tolerated statin therapy, or
-
Bile acid sequestrants, or
-
Ezetimibe, or
-
Niacin
-
Estrogen replacement therapy in postmenopausal women
Consider LDL apheresis for the following patients:
-
Those with documented CHD whose LDLc level cannot be lowered below 200 mg/dL by conventional therapy
-
Those without CHD but who have an LDLc level greater than 300 mg/dL
The 2013 EAS consensus statement for treatment of heterozygous FH includes the following recommendations [4, 5] :
-
An LDL target of < 3.5 mmol/L (< 135 mg/dL) for children with FH (age 8–10);
-
An LDL target of < 2.5 mmol/L (< 100 mg/dL) for adults with FH; and
-
An LDL target of < 1.8 mmol/L (< 70 mg/dL) for adults with known CHD or diabetes.
See Treatment and Medication for more details.
Background
Familial hypercholesterolemia (FH) is an autosomal dominant disorder that causes severe elevations in total cholesterol and low-density lipoprotein cholesterol (LDLc). [1, 2, 3] Although moderate hypercholesterolemia is a common finding in industrialized countries, heterozygous FH occurs in approximately 1 in 200-250 persons in the general population, about two-fold higher than previously thought. [13]
Because FH is associated with a high risk for premature coronary artery disease (CAD), health professionals should be alert to the signs found during a physical examination and to the laboratory values suggestive of FH. [14] Early detection and aggressive management to lower the LDLc level helps prevent or slows the progression of coronary atherosclerosis. Moreover, if the first-degree relatives of a patient with FH are screened, other gene carriers can be identified and treated. [15]
Pathophysiology
FH is a disorder of absent or grossly malfunctioning low-density lipoprotein (LDL) receptors. The LDL receptor gene is located on the short arm of chromosome 19, and the protein is composed of 860 amino acids. It is the primary determinant of hepatic LDL uptake, which normally processes approximately 70% of circulating LDL. Two ligands on LDL bind to the receptor, apolipoprotein B-100 (apoB-100) and apoE. The LDL receptor also binds another ligand, apoE, and is, therefore, more accurately termed the B,E receptor. ApoE is found on most lipoproteins other than LDL, including very low-density lipoprotein (VLDL) and chylomicrons and their remnants, intermediate-density lipoprotein (IDL), and a subclass of high-density lipoprotein (HDL). The LDL receptor binds apoE with higher affinity than apoB-100, and some mutations in the receptor may spare uptake of LDL by allowing binding to apoE. [16, 17, 18]
Goldstein and Brown discovered the LDL receptor and determined that FH was caused by an autosomal dominant mutation. [19, 20] Since then, more than 1700 mutations have been identified, with 79% of of them probably expressed as a hypercholesterolemic phenotype. Defects in the genes encoding apoB and proprotein convertase subtilisin/kexin type 9 (PCSK9) are responsible for approximately 5% and less than 1% of FH cases, respectively. [13] LDL receptor function varies from nonexistent up to about 25% of normal receptor activity. [21]
Five classes of mutations have been defined as follows:
-
Class 1 includes null alleles that result in complete absence of the LDL receptor.
-
Class 2 includes defective transport alleles, which disrupt normal folding of the receptor and cause either failure in transport to the cell surface or successful transport of truncated, mutated receptors.
Class 2a mutations completely block the transport of the receptor from the endoplasmic reticulum to the Golgi apparatus.
Class 2b mutations result in a partial blockade of transport of the receptor from the endoplasmic reticulum to the Golgi apparatus.
-
Class 3 includes defective binding alleles that affect binding of LDL and, in some cases, binding of VLDL as well.
-
Class 4 includes defective internalization alleles that affect the concentration of normal receptors in clathrin-coated pits for internalization by the hepatocyte.
-
Class 5 includes defective recycling alleles that prevent dissociation of the receptor and the ligand and thereby interrupt recycling of the receptor.
Frequency
United States
The prevalence of heterozygous FH has been thought to be approximately 1 case per 500 persons, although it has been more recently estimated at 1 case per 299 persons. [22] The prevalence of homozygous FH is 1 case per 1 million persons.
International
The prevalence of heterozygous FH in Europe approximates that of the United States, but certain regions, such as Iceland and Finland, or populations have a higher incidence. The prevalence of heterozygous FH among French Canadians is 1 case per 270 persons and is 1 case per 170 persons in Christian Lebanese. Due to the founder effect and relatively isolated populations, 3 distinct populations within South Africa have an extremely high prevalence of FH: 1 case per 67 in Ashkenazi Jews and 1 case per 100 persons in both Afrikaners and South African Indians.
Mortality/Morbidity
Homozygous FH
Severe and widespread atherosclerosis affects all major arterial beds, including the carotid, coronary, femoral, and iliac.
Children are at risk for early coronary events, and sudden death or acute myocardial infarction may occur in patients as young as 1-2 years. Without heroic interventions to lower blood cholesterol levels, survival beyond young adulthood is unlikely.
Valve abnormalities are common, particularly aortic stenosis.
Accumulation of cholesterol in nonvascular tissue is of less clinical significance. Corneal arcus and planar, tendon, and tuberous xanthomas are present early in childhood and sometimes at birth. Recognition of the cutaneous manifestations of FH permits early diagnosis and treatment to prevent the otherwise severe and inevitable cardiovascular complications. [23, 24]
Heterozygous FH
Premature CAD is the most serious and preventable manifestation. Untreated men are likely to develop symptoms by the fourth decade of life. The onset of symptoms in women lags behind men by approximately 10-15 years. No accurate estimates of mortality rates are available.
Cholesterol deposition in nonvascular tissue is common, although heterozygous children do not usually have physical manifestations; adults do not invariably develop them. Corneal arcus is the most frequent finding, particularly in patients older than 30 years, but this finding is also common in older patients and African Americans without hypercholesterolemia. Similarly, xanthelasmas (palpebral xanthomas) can occur in older individuals with normal cholesterol levels. Neither xanthelasma nor corneal arcus is of clinical significance, except possibly cosmetically.
Xanthomas, most commonly of the Achilles tendon and extensor tendons of the hands, are rare in children and common in untreated adults. Tendon xanthomas may occur with other conditions such as familial defective apoB-100 and type III hyperlipoproteinemia. These deposits can cause Achilles tendonitis and articular symptoms, particularly of the hands, wrists, knees, and ankles. [25]
Race
Certain populations with Finnish, Lebanese, Ashkenazi Jewish, Afrikaner, or French Canadian origins have a higher prevalence of FH.
Sex
The gene for FH is on chromosome 19; therefore, the inheritance pattern is the same for males and females.
In heterozygous FH, the consequences of severe hypercholesterolemia manifest earlier in men than in women because of the sex protection that benefits women until the postmenopausal years. Although a woman with no other major risk factors for CAD may not develop symptomatic CAD during her lifetime, men are rarely so fortunate.
Homozygous girls and boys have the same risk for a very early cardiovascular event.
Age
The consequences of a defective LDL receptor and subsequent elevations of LDLc are present at birth, but age is relevant because the longer patients live with extremely elevated LDLc levels, the higher their risk of CAD.
Early diagnosis and treatment to lower LDL levels and treat other coronary risk factors slows the progression of coronary atherosclerosis.
-
Metacarpophalangeal joint tendon xanthomas in a 45-year-old man with heterozygous familial hypercholesterolemia.
Tables
Risk Category |
LDLc Target level, mg/dL |
LDLc level Indicating TLC, mg/dL |
LDLc level for Considering Drug Therapy, mg/dL* |
High risk: CHD or CHD risk equivalent (10-y risk >20%) |
< 100 Optional goal < 70 |
>100 |
>100 |
Moderately high risk: More than 2 risk factors (10-y risk 10-20%) |
130 Optional goal < 100 |
>130 |
>130 (100-129 may consider drug options) |
Moderate risk: More than 2 risk factors (10-y risk 10%) |
< 130 |
>130 |
>160 |
Lower risk: 0-1 risk factor |
< 160 |
>160 |
>190 (160-189 LDL-lowering drug optional) |
*The 2004 update recommended that when statin therapy is initiated in patients at high or moderately high risk, a dose and strength should be chosen that achieves at least a 30-40% LDLc reduction (see Table 3). |
|||
Food Category |
Typical US Diet |
NCEP Diet |
Diet for FH |
Cholesterol, mg/d |
500 |
< 200 |
100 |
Total fat, % energy (calories) |
40 |
25-35 |
20 |
Saturated fat, % energy (calories) |
14 |
< 7 |
< 6 |
Carbohydrate, % energy (calories) |
45 |
50-60 |
65 |
Protein, % energy (calories) |
Approximately 15 |
15 |
N/A |
Statin |
FDA-Approved Dose |
Expected LDLc Decrease |
Dose Required for 30-40% LDLc Reduction |
Atorvastatin |
10-80 mg daily |
35-60% |
10 mg |
Fluvastatin |
20-40 mg at bedtime |
20-30% |
40 mg qd/bid |
40 mg bid |
35% |
40 mg bid |
|
Extended-release fluvastatin (Lescol XL) |
80 mg at bedtime |
35-38% |
80 mg at bedtime |
Lovastatin |
20-80 mg at supper |
25-48% |
40 mg at dinner |
Extended-release lovastatin (Altoprev) |
20-60 mg at bedtime |
25-45% |
60 mg at bedtime |
Pravastatin |
40-80 mg at bedtime |
30-40% |
40 mg at bedtime |
Rosuvastatin |
10-40 mg daily |
40-60% |
5 mg daily |
Simvastatin |
20-80 mg daily at bedtime |
35-50% |
20 mg at bedtime |
Simvastatin + ezetimibe (Vytorin) |
10/20 mg 10/40 mg 10/80 mg at bedtime |
50-60% |
10/20 mg at bedtime |





