Background
Hirschsprung disease is a developmental disorder characterized by the absence of ganglia in the distal colon, resulting in a functional obstruction. [1] See the images below.
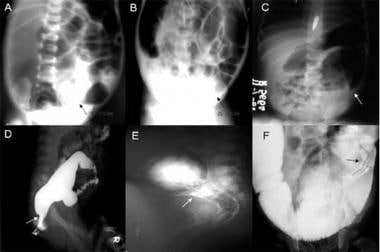 A: Plain abdominal radiograph showing a transition zone (PARTZ) at the rectosigmoid. B: Plain abdominal radiograph showing a PARTZ at the midsigmoid. C: Plain abdominal radiograph showing a PARTZ at the descending colon. D: Contrast enema showing a contrast enema transition zone (CETZ) at the rectosigmoid. E: Contrast enema showing a CETZ at the midsigmoid. F: Contrast enema showing a CETZ at descending colon. Images courtesy of Pratap A, Gupta DK, Tiwari A, et al. BMC Pediatr. 2007 Jan 27;7:5. [Open access.] PMID: 17257439, PMCID: PMC1790893.
A: Plain abdominal radiograph showing a transition zone (PARTZ) at the rectosigmoid. B: Plain abdominal radiograph showing a PARTZ at the midsigmoid. C: Plain abdominal radiograph showing a PARTZ at the descending colon. D: Contrast enema showing a contrast enema transition zone (CETZ) at the rectosigmoid. E: Contrast enema showing a CETZ at the midsigmoid. F: Contrast enema showing a CETZ at descending colon. Images courtesy of Pratap A, Gupta DK, Tiwari A, et al. BMC Pediatr. 2007 Jan 27;7:5. [Open access.] PMID: 17257439, PMCID: PMC1790893.
Most cases of Hirschsprung disease are diagnosed in the newborn period. Hirschsprung disease should be considered in any newborn who fails to pass meconium within 24-48 hours of birth. Although contrast enema is useful in establishing the diagnosis, full-thickness rectal biopsy remains the criterion standard. Once the diagnosis is confirmed, the definitive treatment is to remove the aganglionic bowel and to restore continuity of the healthy bowel with the distal rectum, with or without an initial intestinal diversion.
Historical notes
Although this condition was described by Ruysch in 1691 and popularized by Hirschsprung in 1886, the pathophysiology was not clearly determined until the middle of the 20th century, when Whitehouse and Kernohan reported aganglionosis of the distal colon as the cause of obstruction in a case series. [2]
In 1949, Swenson described the first consistent definitive procedure for Hirschsprung disease, rectosigmoidectomy with coloanal anastomosis. Since then, other operations have been described, including the Duhamel and Soave techniques. More recently, earlier diagnosis and advances in surgical techniques have resulted in decreased morbidity and mortality in patients with Hirschsprung disease.
Also see Pediatric Hirschsprung Disease and Hirschsprung Disease Imaging.
Pathophysiology
Three nerve plexuses innervate the intestine: the submucosal (Meissner) plexus, the myenteric (Auerbach) plexus (between the longitudinal and circular muscle layers), and the smaller mucosal plexus. All of these plexuses are finely integrated and are involved in all aspects of bowel function, including absorption, secretion, motility, and blood-flow regulation.
Normal motility is primarily under the control of intrinsic neurons. In the absence of extrinsic signals, bowel function remains adequate, owing to the complex reflexive architecture of the enteric nervous system (ENS). For this reason, the ENS is often referred to as the “second brain.” Intestinal smooth muscle contraction and relaxation are under the control of enteric ganglia. Most enteric nervous activation causes muscle relaxation, mediated by nitric oxide and other enteric neurotransmitters. Extrinsic neural afferents to the ENS contain cholinergic and adrenergic fibers. The cholinergic fibers generally cause contraction, whereas the adrenergic fibers mainly cause inhibition.
In patients with Hirschsprung disease, both myenteric and submucosal plexuses are absent. The anus is invariably affected, and aganglionosis continues proximally for a variable distance. In the absence of ENS reflexes, control of the intestinal smooth muscle is overwhelmingly extrinsic. The activity of both the cholinergic system and the adrenergic system is 2-3 times that of normal intestine. The cholinergic (excitatory) system is thought to predominate over the adrenergic (inhibitory) system, leading to an increase in smooth muscle tone. With the loss of the intrinsic enteric relaxing impulses, the increased muscle tone is unopposed. This phenomenon leads to an imbalance of smooth muscle contractility, uncoordinated peristalsis, and a functional obstruction.
Enteric ganglion cells are derived from the neural crest during embryonic development. In normal development, the neuroblasts are found in the esophagus by the fifth week of gestation, and they migrate to the small intestine by the seventh week and to the colon by the twelfth week. [3] One possible etiology of Hirschsprung disease is the arrest of aboral neuroblast migration. Alternatively, although normal cell migration may occur, neuroblasts may be subject to apoptosis, failure of proliferation, or improper differentiation within the affected distal intestinal segment. Fibronectin, laminin, neural cell adhesion molecule (NCAM), and neurotrophic factors present in the intestinal stroma are necessary for normal enteric ganglion development, whereas their absence or dysfunction may also have a role in the etiology of Hirschsprung disease. [4, 5, 6]
Investigators have also identified several genes whose improper expression results in a Hirschsprung disease phenotype. Genome-wide association studies (GWAS) in Europeans and Asians have identified three common disease-susceptibility variants at the RET, SEMA3, and NRG1 loci. [7] Rs80227144 is a low-frequency variant of SEMA3 that has been associated with Europeans, and a conditional analysis indicates that rs9282834, low-frequency missense variant encoding RET p.Asp489Asn, is specific to Asians. [7]
The RET protooncogene has been implicated in several studies of Hirschsprung pathogenesis.
So and colleagues discovered that rare variants of RET were associated with more severe phenotypes among Chinese Hirschsprung patients. [8] Leon and colleagues determined that sporadic RET coding sequence mutations in Hirschsprung patients resulted in protein truncations that would deter cell membrane translocation and anchoring. [9]
Qin and colleagues performed microarray analyses of aganglionic colon and normal tissue; they discovered 622 genes with anomalous expression in the aganglionic tissue, and myenteric HAND2 expression was significantly attenuated. [10]
In a comparison of gene expression among normal and aganglionic colon, Chen and colleagues determined that overexpression of DVL1 and DVL3 genes was associated with the Hirschsprung phenotype. [11]
In a review, Butler Tjaden and Trainor reported that mutations in the genes, RET, GDNF, GFRα1, NRTN, EDNRB, ET3, ZFHX1B, PHOX2b, SOX10, and SHH are present in approximately 50% of Hirschsprung disease patients. [12]
These studies indicate the complexity of Hirschsprung pathogenesis. Ongoing studies of genetic and environmental factors will continue to elucidate this problematic disease in the future.
Although enteric ganglion cells are the primary pathogenic entity in Hirschsprung disease, some studies suggest that other cell types may also be implicated. [13, 14, 15, 16, 17] When extrinsically stimulated, smooth muscle cells in aganglionic colon are electrically inactive. [13] Furthermore, interstitial cells of Cajal, pacemaker cells connecting enteric nerves and intestinal smooth muscle, have also been postulated as an important contributing factor. [14, 15, 16] These findings suggest that Hirschsprung pathophysiology is not limited to cells normally present within the enteric ganglia alone.
Epidemiology
United States statistics
Hirschsprung disease affects approximately 1 case per 5400-7200 newborns annually.
International statistics
Although the exact worldwide incidence is unknown, international studies have reported rates ranging from approximately 1 case per 1500-7000 newborns. [18, 19]
Race-, sex-, and age-related demographics
Hirschsprung disease affects all races; however, it is roughly 3 times more common among Asian-Americans. [20]
This disease occurs more often in males than in females, with a male-to-female ratio of approximately 4:1; however, the ratio in long-segment disease decreases to 2:1.
Hirschsprung disease is uncommon in premature infants. However, the age at which Hirschsprung disease is diagnosed has progressively decreased over the past century. In the early 1900s, the median age at diagnosis was 2-3 years; from the 1950s to 1970s, the median age was 2-6 months. Currently, approximately 90% of patients with Hirschsprung disease are diagnosed in the newborn period. [21]
Prognosis
Reports of long-term outcomes after definitive repair for Hirschsprung disease are conflicting. Some investigators report a high degree of satisfaction, whereas others report a significant incidence of constipation and incontinence. In general, more than 90% of patients with Hirschsprung disease report satisfactory outcomes; however, many patients experience disturbances of bowel function for several years before normal continence is established. Approximately 1% of patients with Hirschsprung disease have debilitating incontinence requiring a permanent colostomy.
Total colonic aganglionosis is associated with a poorer outcome, with 33% of patients experiencing persistent incontinence and 14% requiring a permanent ileostomy. Patients with associated chromosomal abnormalities and syndromes also have poorer clinical outcomes.
Morbidity/mortality
Hirschsprung disease is confined to the rectosigmoid region in about 75% of cases. Approximately 60% of infants with Hirschsprung disease have an associated condition, ranging from subtle to severe. Ophthalmologic problems affect 43% of infants, 20% have congenital anomalies of the genitourinary tract, 5% have congenital heart disease, 5% have hearing impairment, and 2% have central nervous system anomalies. [22, 23]
Hirschsprung disease is associated with chromosomal abnormalities or syndromes in approximately 9% of cases. [23] It may be associated with the following syndromes:
-
Down syndrome: Trisomy 21 is characterized by physical growth delays, characteristic facial features, and varying degrees of intellectual disability.
-
Neurocristopathy syndromes: Neurocristopathies represent a broad range of disorders characterized by aberrant neural crest development.
-
Waardenburg-Shah syndrome: Patients have a sensorineural hearing deficit and hypopigmentation of the iris and hair.
-
Yemenite deaf-blind syndrome: Affected patients have a mutation in the SRY-related HMG-box gene. The syndrome is characterized by a severe hearing deficit, nystagmus, patchy pigmentation abnormalities, and defects of the iris and cornea.
-
Piebaldism: Patients have patches of hypopigmentation due to a congenital disorder of melanocytes. Underlying pathology in the neural crest development is also common in Hirschsprung disease.
-
Multiple endocrine neoplasia type II (MEN2): Mutations in the RET protooncogene are associated with medullary thyroid cancer in MEN2 and Hirschsprung disease.
-
Congenital central hypoventilation syndrome (CCHS): CCHS is characterized by absent autonomic control of ventilation. PHOX2B transcription factor mutations may be associated in both CCHS and Hirschsprung disease.
Untreated aganglionic megacolon in infancy may result in a mortality rate as high as 80%. Operative mortality rates for any of the interventional procedures are very low. Even in cases of treated Hirschsprung disease, the mortality rate may approach 30% as a result of severe enterocolitis.
Possible complications of surgery include anastomotic leak (5%), anastomotic stricture (5%-10%), intestinal obstruction (5%), pelvic abscess (5%), and wound infection (10%). Long-term complications mostly affect patients with long-segment disease. These include chronic obstructive symptoms, incontinence, chronic constipation, enterocolitis, and late mortality. Although many patients encounter one or more of these problems postoperatively, long-term follow-up studies have shown that greater than 90% of children experience significant improvement. [24] Patients with a syndromic association and those with long-segment disease have poorer outcomes. [25, 26, 27]
Complications
Potential complications for the complex operations associated with Hirschsprung disease encompass the entire spectrum of gastrointestinal surgical complications. The incidence rates of these complications do not appear to vary significantly with the surgeon’s experience.
The most frequent postoperative complications include enterocolitis after the Swenson procedure, constipation following the Duhamel repair, and diarrhea and incontinence after the Soave pull-through procedure.
Overall, the most common complications are anastomotic leakage and stricture formation in 5%-15%, wound infection in 10%, intestinal obstruction in 5%, pelvic abscess in 5%, and reoperation in 5% of patients. After intestinal diversion, patients may also develop enterostomal complications, such as prolapse, herniation, or stricture.
In a retrospective (1979-2016) study of medical chart information from 657 Korean patients with Hirschsprung disease who underwent pull-through surgeries, investigators found 49 patients (7.5%) had redo pull-through procedures. [28] Causes of redo included pathologic problems (aganglionosis, hypoganglionosis, immature ganglion cell) and anatomic problems (stricture, fistula and/or abscess at anastomosis), with those who had anatomic problems undergoing third redo procedures more than the group with pathologic problems. [28]
Enterocolitis, chronic obstruction, incontinence, constipation, and late mortality may occur late after surgery. Rectovesical fistulas have also been reported in the literature. [29]
Enterocolitis accounts for significant morbidity and mortality in patients with Hirschsprung disease and can progress into toxic megacolon. [30] Enterocolitis is characterized by inflammation of the colon or the small intestinal mucosa. As the disease progresses, the intestinal lumen fills with a fibrinous exudate, and the risk of perforation increases. This process may occur in either aganglionic or ganglionic segments of the bowel. Patients typically present with explosive diarrhea, abdominal distention, fever, emesis, and lethargy. Approximately 10%-30% of patients with Hirschsprung disease develop enterocolitis. Long-segment disease is associated with an increased incidence of enterocolitis. The risk of enterocolitis does not decrease with surgical correction.
Patients may present postoperatively with abdominal distention, emesis, or constipation indicative of ongoing obstruction. [31] Mechanical obstruction can be diagnosed with digital rectal examination and barium enema. Serial anorectal dilatations or surgical revision of the pull-through may be required. [4]
Persistent aganglionosis occurs rarely and may be due to pathologic error, inadequate resection, or loss of ganglion cells after the pull-through procedure. [32] If a rectal biopsy does not show ganglion cells, revision of the pull-through must be done. [33, 34, 35, 36]
Hirschsprung disease may be associated with other disorders of intestinal motility. Contrast studies, manometry, and biopsy may be necessary to evaluate for intestinal neuronal dysplasia. [37, 38]
Internal sphincter achalasia may result in persistent obstruction. This can be treated with internal sphincterotomy, intrasphincteric botulinum toxin, or nitroglycerin paste. Most cases resolve by age 5 years. [39, 40]
Functional megacolon may be present due to stool-holding behavior. Bowel management regimens may be implemented with cecostomy and antegrade enemas reserved for refractory cases. [41]
Incontinence may be the result of abnormal sphincter function, decreased sensation, or overflow incontinence secondary to constipation. [31] Anorectal manometry and ultrasonography are useful to distinguish between these entities.
-
A: Plain abdominal radiograph showing a transition zone (PARTZ) at the rectosigmoid. B: Plain abdominal radiograph showing a PARTZ at the midsigmoid. C: Plain abdominal radiograph showing a PARTZ at the descending colon. D: Contrast enema showing a contrast enema transition zone (CETZ) at the rectosigmoid. E: Contrast enema showing a CETZ at the midsigmoid. F: Contrast enema showing a CETZ at descending colon. Images courtesy of Pratap A, Gupta DK, Tiwari A, et al. BMC Pediatr. 2007 Jan 27;7:5. [Open access.] PMID: 17257439, PMCID: PMC1790893.
-
Hirschsprung disease. Contrast enema demonstrating transition zone in the rectosigmoid region.









