Overview
A wide variety of immunologic, idiopathic, neoplastic, infectious, and fungal processes can cause a chronic granulomatous reaction in the head and the neck (see the images below). A granuloma is a focal area of chronic inflammation produced by circulating monocytes as part of an immunologic process. A granulomatous reaction is characterized histologically by transformed macrophages (epithelioid cells) surrounded by lymphocytes. These epithelioid cells may later fuse to form giant cells.
Granulomatous diseases cause a great range of symptoms not only in the head and neck but also throughout the body. Because patients initially present to the otolaryngologist's office, a full history and a complete physical examination are emphasized for all patients. Although similar histologically, granulomatous diseases of the head and the neck require a thorough approach because of the wide variety of disease processes.
Autoimmune Granulomatous Diseases
Granulomatosis with polyangiitis (Wegener granulomatosis)
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a disease that typically consists of a triad of airway necrotizing granulomas, systemic vasculitis, and focal glomerulonephritis. If the disease does not involve the kidneys, it is called limited granulomatosis with polyangiitis. Although the cause of granulomatosis with polyangiitis is still not certain, it is believed to most likely have an autoimmune etiology. [1, 2, 3, 4, 5, 6]
Symptoms
Granulomatosis with polyangiitis most commonly occurs in whites during the third to fifth decades of life. Often, the patient initially presents with symptoms that involve the head and neck. Inflamed, friable mucosa; ulcerations; septal perforations; and saddle nose deformities may be seen on nasal examination; therefore, the patient may present with various nasal symptoms, including chronic nasal congestion, epistaxis, and pain.
The patient may also present with laryngeal symptoms (wheezing), ear symptoms (otitis media), and oral ulcerations. Pulmonary symptoms include cough and hemoptysis. A mismatch often occurs in the severity of the symptoms and the relatively benign appearance of the lesions.
With regard to cutaneous lesions, a study by Montero-Vilchez et al found that out of 21 patients with granulomatosis with polyangiitis, the chief sites of these were the lower limbs (57.14%) and the face (23.81%). [7]
Laboratory examination
Laboratory evaluation includes elevated inflammatory indicators, such as C-reactive protein and erythrocyte sedimentation rate (ESR). An elevated classic antineutrophil cytoplasmic antibody (C-ANCA) level is 80% specific for granulomatosis with polyangiitis. In disseminated disease, 78-100% sensitivity is reported, whereas in upper airway disease, 60-70% sensitivity is reported.
Histologic examination reveals granulomatous change consisting of macrophages and accompanying inflammatory cells and giant cells. A characteristic patchy necrosis surrounded by giant cells through the tissue is often present. The vasculitis reveals granulomatous inflammation to the vessel wall in small and medium-sized arteries and veins, making granulomatosis with polyangiitis a true vasculitis. Other features that may be noted on histopathologic examination include microabscess formation and cicatricial scarring.
Treatment
Treatment includes steroids for 4-6 weeks until the disease is controlled. Steroids are then tapered over 6 months. Cyclophosphamide is an alkylating chemotherapeutic agent that can be tapered to a maintenance dose. Trimethoprim-sulfamethoxazole has also demonstrated early promise.
A study by Puéchal et al indicated that azathioprine and methotrexate have comparable efficacy in maintaining remission in granulomatosis with polyangiitis. The 10-year overall survival rate for patients receiving azathioprine was 75.1%, compared with 79.9% for those receiving methotrexate, while the relapse-free survival rate was 26.3% for patients receiving azathioprine, compared with 33.5% for those receiving methotrexate. [8]
A study by Springer et al indicated that discontinuation of or use of low drug dosages in maintenance therapy increases the risk of relapse in granulomatosis with polyangiitis. The study involved 157 patients with the disease who, after achieving remission, underwent maintenance therapy with methotrexate or azathioprine; they were followed up for a median period of 3.1 years. In patients who received maintenance medication for more than 18 months, the hazard ratio (HR) for relapse was reduced by 29%, while in those who continued maintenance treatment for more than 36 months, the HR for relapse was reduced by 66%. Fifty-two percent of relapses took place in patients who were no longer on maintenance therapy. Among patients on methotrexate, 52% who relapsed during therapy were receiving less than 15 mg per week, while among those on azathioprine, 67% of those who relapsed during treatment were receiving 50 mg or less per day. [9]
Repeated inflammatory insult in granulomatosis with polyangiitis leads to focal areas of scarring and fibrotic damage to airway structures. Only 20% of cases of subglottic stenosis due to the disease will resolve with immunotherapy. Therefore, laser treatment, tracheal resection and anastomosis, and tracheotomy are treatment options for subglottic stenosis secondary to granulomatosis with polyangiitis. The prognosis is good if the disease is treated early and prior to significant renal involvement.
Relapsing polychondritis
Relapsing polychondritis is a rare autoimmune disorder that affects cartilage and tissue containing glycosaminoglycans. It often presents with other autoimmune disorders.
Typical manifestations include nasal chondritis (15%), auricle chondritis (50%), nonerosive polyarthritis (50%), respiratory tract chondritis (15%), and ocular inflammation (15%). This recurrent chondritis presents with sudden, painful episodes that resolve in 7 days. The sequelae include saddle nose deformity, auricular deformity, airway compromise, and visual disturbance.
Although gross findings are nonspecific, relapsing polychondritis is characterized by an overall thickening of the cartilage in the head and the neck, including the ear, the nose, the epiglottis, and the cricoid and tracheal rings. The perichondrium is infiltrated by a spectrum of inflammatory cells, leading to its destruction.
Laboratory evaluation reveals an elevated erythrocyte sedimentation rate (ESR) level during disease activity. The most specific test available looks at antibodies to collagen type II, which is exclusively found in cartilage. Treatment includes immunosuppressive agents. Steroids are most commonly used.
Lupus
Lupus is a disease that is caused by the deposition of antibodies and immune complexes (type III hypersensitivity). It typically occurs in young black female patients. Patients with human leukocyte antigen DR2 (HLA-DR2) and HLA-DR3 have a genetic predisposition to lupus. Three subtypes of lupus exist: discoid lupus erythematosus, subacute cutaneous lupus, and systemic lupus erythematosus
Discoid lupus erythematosus
Discoid lupus erythematosus is the least aggressive form and affects only superficial tissues. [10] Patients with discoid lupus erythematosus may present with oral or cutaneous lesions consisting of elevated erythematous plaques, hypopigmented edges, and alopecia. Scarring from these lesions may be the only sequela. Diagnosis is based on clinical examination because the findings from the antinuclear antibody (ANA) test and the lupus erythematosus (LE) cell test are negative.
A study by Yavuz et al of 132 individuals with discoid lupus erythematosus indicated that smoking and chronic exposure to ultraviolet (UV) rays are risk factors for the disease, having been found in 61.4% and 42.4% of the patients, respectively. [11]
Subacute cutaneous lupus
Subacute cutaneous lupus presents with papulosquamous lesions that do not scar. Mild systemic involvement may occur. The findings from the ANA test and the Sjögren syndrome A antibody (SS-A [anti-Ro]) and Sjögren syndrome B antibody (SS-B [anti-La]) tests are inconsistent and cannot be used to aid in making a diagnosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is the most severe form of lupus. Typically, patients may present with a malar butterfly rash. [12] Multiple visceral organs may be involved. Laryngeal involvement includes thickening of the true vocal cords and perichondritis of the laryngeal and tracheal cartilages that may present as hoarseness and pain. Anterior septal perforations are not uncommon and are a result of crusting and nasal dryness. [13]
The ANA test is 98% specific for systemic lupus erythematosus, and the findings from the LE cell test and the SS-A (anti-Ro) and SS-B (anti-La) tests are typically positive. A nonspecific test, such as the ANA test, the SS-A (anti-Ro) test, or the SS-B (anti-La) test, is initially used to aid in establishing a diagnosis. Specific markers include anti–double-stranded (DS) DNA and Sm antigen (SmAg). Treatment typically includes nonsteroidal anti-inflammatory drugs and steroids.
Behçet disease
Behçet disease is a rare disorder that consists of recurrent aphthous ulcers on the oral mucosa and the genitalia. [13] Ocular inflammation is also present. [14] A study by Gueudry found that interferon (IFN)-alpha2a is effective and safe in the long-term therapeutic treatment of severe uveitis. [15] One report suggests the incidence of sensorineural hearing loss was significantly higher than in controls, and an audiogram is suggested. [16]
Allergic granulomatosis and vasculitis (Churg-Strauss syndrome)
Churg-Strauss syndrome is characterized by the triad of asthma, systemic vasculitis, and eosinophilia (peripherally and within the tissue). Patients with Churg-Strauss syndrome may present with nasal symptoms similar to those of granulomatosis with polyangiitis. (A study by Fina et al of 14 children with Churg-Strauss syndrome found that 85% had upper airway involvement. [17] ) Histologic examination reveals granulomatous changes to the tissue, and true vasculitis and eosinophilia are present.
Granulomatous Diseases of Unknown Etiology
Sarcoidosis
Sarcoidosis is an idiopathic, systemic granulomatous disease of unknown etiology with a predilection for the lungs and the upper respiratory tract. Although the cause of sarcoidosis is not known, it may be due to an immunologic disorder. [18, 19]
The disease predominates in females, with a peak incidence in the third to fifth decades of life. Sarcoidosis is 10-20 times more common in blacks in North America, whereas blacks on other continents are spared.
The disorder most commonly presents with hilar adenopathy detected on an incidental chest radiograph. Almost 90% of patients with sarcoidosis have pulmonary involvement that can cause cough, hilar adenopathy, and dyspnea. In the larynx, sarcoidosis typically presents as a submucosal mass in the supraglottis, most commonly in the epiglottis. True vocal fold paralysis has been reported. Nasal examination may reveal cobblestoning of sinonasal mucosa, crusting, and epistaxis.
Krespi described 3 stages of sinonasal sarcoidosis, as follows:
-
Stage I - Limited reversible involvement of the paranasal sinuses
-
Stage II - Moderate disease involvement or limited single sinus involvement.
-
Stage III - Irreversible disease causing intranasal synechiae, nasal stenosis, and cartilage destruction
Braun and associates found that the most common presentation was of chronic, nondestructive, crusty, inflammatory nodules on the septum and turbinates. Functional endoscopic sinus surgery (FESS) can improve a patient's quality of life, but recurrence is likely.
Sarcoidosis may affect the salivary glands, and its presentation may range from an asymptomatic parotid mass to Heerfordt disease, an extrapulmonary sarcoid manifestation of uveitis, facial palsy, sensorineural hearing loss, fever, and parotid enlargement. The disease may cause a progressive interstitial lung disease, and blindness may result from the progression of uveitis. Other cutaneous findings include erythema nodosum and Darier-Roussy nodule (subcutaneous nodules). Cardiac arrhythmias, neuropathies, and hepatic and renal involvement may also be present.
A study by Hofauer et al indicated that in patients with sarcoidosis, those in whom initial symptoms, such as swelling or pain, are present in the area of the salivary glands are more likely to experience dry mouth and eyes. However, these symptoms regressed in all patients in the study in whom they manifested. Permanent sarcoidosis-associated abnormalities in the salivary gland region appear to occur only in individual cases. [20]
Diagnosis and treatment
Establishing a diagnosis is complex, and evidence can consist of findings on laboratory studies, biopsy specimens, and radiologic studies. The Kveim-Siltzbach skin test involves an intradermal injection of an antigen extract from a patient who is known to have this disease. If the patient develops a nodule within 4-8 weeks and if histopathologic examination reveals noncaseating granulomas, the likelihood of disease is 80%. Elevated angiotensin-converting enzyme (ACE) levels and urine/serum calcium levels may also suggest sarcoidosis.
According to Siltzbach, chest radiographic findings can be divided into 4 stages, as follows:
-
Stage I - Consists of bilateral hilar lymphadenopathy
-
Stage II - Consists of bilateral hilar lymphadenopathy and pulmonary infiltrates
-
Stage III - Consists of pulmonary infiltrates alone
-
Stage IV - Consists of fibrosis and honeycombing of the lung fields
Histologic examination reveals a granulomatous disease consisting of discrete, noncaseating epithelioid granulomas; an accumulation of T cells; mononuclear macrophages; and giant cells. The giant cells associated with sarcoidosis are usually larger than those associated with tuberculosis, and intracytoplasmic inclusion bodies may be present.
Treatment includes steroids for exacerbations of the pulmonary disease. Isolated sinus lesions may be treated with topical and injectable steroids. Some authors have reported good success with endoscopic sinus surgery. [21] Supraglottic lesions are usually monitored clinically, with surgery reserved for obstructing lesions.
Idiopathic midline destructive disease
As its name implies, idiopathic midline destructive disease (IMDD) is a midline destructive disease. The disease is localized to the head and the neck, and it may present with pansinusitis and ulceration of the nasal floor and septal ulcerations. Biopsy specimens reveal sheets of typical polymorphonuclear cells without granulomas or vasculitis. Studies from Asian and Western countries have shown a possible association with Epstein-Barr virus.
The lack of granulomatous change on histologic examination makes the classification of IMDD as a granulomatous disease controversial. Treatment consists of radiation therapy for local disease; cyclophosphamide and prednisone are administered for multiregional disease.
Neoplastic Granulomatous Diseases
Histiocytosis X (Langerhans cell histiocytosis)
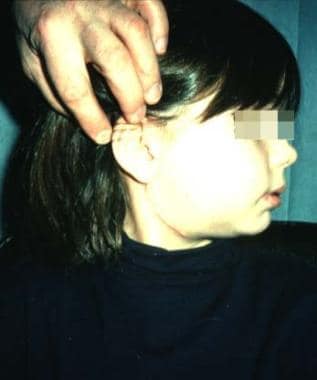
The classification of histiocytoses is confusing and not well established. Some of the nomenclature refers to this group of diseases as Langerhans cell histiocytosis. Overall, some observations on histiocytoses can be made, as follows (see the image below) [22, 23] :
-
Histiocytoses most commonly present as otitis media in patients
-
On anterior nasal rhinoscopy, a nasal mass, epistaxis, or septal perforation may be seen
-
Histologic examination reveals sheets of polygonal histiocytes with a variable number of eosinophils, plasma cells, and lymphocytes
-
The nucleus of the Langerhans cells is characteristically pale and deeply grooved
-
The cytoplasm of the Langerhans cells may demonstrate Birbeck granules on electron microscopy
-
The prognosis is poor for patients who are young at the time of presentation
Eosinophilic granuloma
Eosinophilic granuloma is the localized form of histiocytosis X occurring in children and young adults. The disease may be monostotic or polyostotic, typically affecting the temporal bones and the frontal bones. The presentation may vary and includes acute mastoiditis, middle ear granulation tissue, tympanic membrane perforations, proptosis, and facial nerve paralysis. The histologic characteristics of eosinophilic granuloma are demonstrated in the image below.
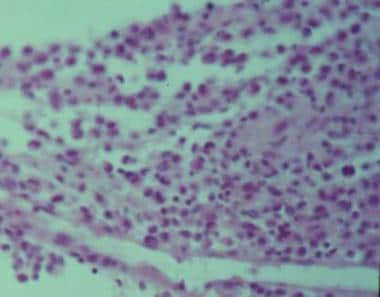
Treatment is typically surgical curettage, with radiation therapy reserved for inaccessible lesions. The prognosis is excellent for patients with eosinophilic granuloma.
Hand-Schüller-Christian disease
This chronic, disseminated form of histocytosis X also occurs in children and young adults. The disease is polyostotic and classically consists of skull lesions, exophthalmos, and diabetes insipidus. Diabetes insipidus results from erosion into the sella turcica.
Treatment of Hand-Schüller-Christian disease typically includes surgical excision, chemotherapy, and radiation therapy. Otitis externa in this disorder may be treated with topical nitrogen mustard. The mortality rate for the Hand-Schüller-Christian disease is approximately 30%.
Letterer-Siwe disease
This disease, an acute, disseminated form of histocytosis X, typically affects infants and young children. Patients with Letterer-Siwe may present with fever, proptosis, splenomegaly, hepatomegaly, or dermatitis. Treatment is a combination of radiation and chemotherapy. This disorder is uniformly fatal.
Fibrous histiocytoma
Fibrous histiocytoma presents with nasal obstruction, epistaxis, dysphagia, and dyspnea. It can occur at any age and is found mostly in males.
Lobular capillary hemangioma (pyogenic granuloma)
Lobular capillary hemangioma, also known as pyogenic granuloma, usually presents as a painless, friable lesion anywhere on the head and neck. It develops in prepubescent males, in postpubescent females, or during pregnancy. It is believed to be hormonally responsive. Treatment is surgical excision.
Infectious Granulomatous Diseases
Catscratch disease
Catscratch disease (CSD) is caused by Bartonella henselae, an intracellular, pleomorphic, gram-negative bacteria. A high percentage of cats carry B henselae.
Patients with CSD typically present with tender regional adenopathy and provide a history of a cat scratch or bite; 50% of patients have a vesicle or a pustule within a week after the cat scratch. Painful lymphadenopathy develops near the primary lesion a few weeks after inoculation. Head and neck lymphadenopathy occurs in about one half of all patients with CSD.
Contact with the cornea (eg, petting the cat and then rubbing the eye) can result in a granulomatous lesion on the eye and preauricular adenopathy (Parinaud oculoglandular syndrome). A retrospective cohort study by Habot-Wilner indicated that among patients with ocular CSD, lesions of the optic nerve head are common, with the investigators having found them in 43 out of 107 CSD-affected eyes (40%). [24]
Warthin-Starry silver stain must be requested to demonstrate the gram-negative bacteria. A polymerase chain reaction (PCR) assay test can also be performed. [25] Biopsy specimens reveal granulomatous changes. Because diagnosis can be difficult, a combination of serologic, epidemiologic, histologic, and molecular criteria should be used. An immunoglobulin-M (IgM) enzyme-linked immunosorbent assay (ELISA) test for detecting an early antibody response to B henselae was found to be 100% sensitive and 97.1% specific. [26]
Treatment includes erythromycin, rifampin, or doxycycline. CSD is fatal if left untreated in patients who are immunocompromised.
Rhinoscleroma
Rhinoscleroma (hard-nosed) is caused by Klebsiella rhinoscleromatis, a gram-negative bacterium. It occurs primarily in Eastern Europe and Central America. [27]
Typically, rhinoscleroma presents with the following 3 stages, which may occur over years:
-
First stage - The catarrhal, or rhinitic, stage presents with purulent rhinorrhea and nasal honeycombed crusting
-
Second stage - The florid, or granulomatous, stage, consists of granulomatous nodules on the head and the neck, especially the glottis and the subglottis.
-
Third stage - The cicatricial stage involves a dense, fibrotic reaction that may eventually stenose the nose, the larynx, the tracheobronchial tree, or a combination of these
Clinical diagnosis is based on the classic honeycomb crusting seen in the nose. Biopsy specimens may reveal pseudoepitheliomatous hyperplasia, Russell bodies, and Mikulicz cells. Russell bodies are bifringent inclusions and bloated plasma cells. Mikulicz cells are multinucleated macrophages that contain the bacteria and are most abundant during the florid phase. Treatment consists of long-term therapy with either streptomycin or tetracycline. If the cicatricial stage causes stenosis, dilatation may be necessary.
Leprosy
Leprosy is caused by exposure to Mycobacterium leprae. Patients may present with hypopigmented macules, tuberculoid skin, mucosal nodules in the mouth and the nose that subsequently ulcerate, laryngeal ulcerations, lymphadenopathy, nasal collapse, and fish-mouth deformity. Treatment consists of dapsone.
Nontuberculous mycobacteria
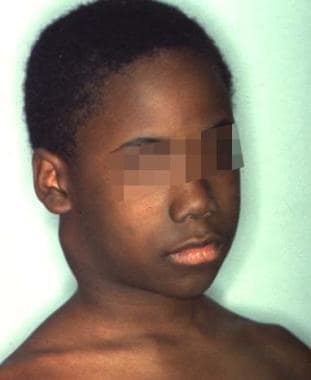
The main organisms responsible for granulomatous change in the head and the neck are Mycobacterium avium-intracellulare complex, M scrofulaceum, M kansasii, M fortuitum, and M bohemicum. Persistent lymphadenitis is the most common cause of nontuberculous infection in children. The route of transmission is from environmental sources (eg, soil in the mouth or the eye).
Head and neck manifestations include corneal ulcerations, cervical lymphadenopathy, and, rarely, mastoiditis. The typical presentation of nontuberculous mycobacteria adenitis is of a slowly enlarging submandibular or preauricular mass in the absence of systemic signs of infection. The mass is typically nontender and does not respond to standard antibiotics. The mass may progress to discoloration and spontaneous drainage through the skin.
Several tests are performed to aid in establishing a diagnosis. Computed tomography (CT) scanning with contrast demonstrates asymmetrical cervical lymphadenopathy, and contiguous, low-density, necrotic, ring-enhancing masses involving subcutaneous fat are typically minimal or absent in nontuberculous mycobacteria adenitis. A significant amount of inflammatory stranding in the subcutaneous fat is not usually seen as in bacterial abscesses.
Diagnosis is by culture and sensitivity, which may take as long as 4 weeks and is reported in approximately 60% of patients. The purified protein derivative (PPD) test is either only mildly reactive or absent. Stains for acid-fast mycobacteria are positive in only 20-50% of patients. A diagnosis may be made empirically based on the pathologic diagnosis, the history, and physical examination findings suggestive of nontuberculous mycobacteria infection.
Treatment involves lymph node excision because incision and drainage may lead to either recurrence or sinus tract formation. In addition to physician experience, antibiotics are chosen by epidemiology and minimum inhibitory concentration (MIC).
Tuberculosis
Tuberculosis, a result of inhalation of M tuberculosis into the lungs, rarely affects the head and neck. Primary infection is typically an asymptomatic lower lobe lesion, and a calcified lung lesion and a draining lymph node are often present, thus forming a Ghon complex. Secondary involvement manifests with night sweats, fever, weight loss, and chronic, nonproductive cough. It also affects apical segments.
The most common manifestation on the head and the neck is cervical lymphadenopathy, presenting as firm and nontender. Tuberculosis appears as nonspecific, homogenous enhancement on CT scan. When present in the larynx, tuberculosis typically involves the posterior glottis, with granulation tissue and ulcerations. Otologically, tuberculosis manifests as a painless, odorless, watery otorrhea with multiple miniature tympanic membrane perforations.
Diagnosis is aided by a PPD test and a chest radiograph. Sputum cultures and biopsy samples may also be obtained.
Actinomycosis
Actinomycosis may result following dental manipulation or trauma and may present as a mass anywhere on the head and the neck. Diagnosis may be challenging and may require surgical excision of the mass. Histologic examination reveals sulfur granules. Treatment is a combination of surgical debridement and penicillin G. [28, 29]
Syphilis
The causative organism of syphilis is Treponema pallidum. Syphilis may present as early stage or late stage. The primary stage presents as a chancre at the inoculation site. The second stage is highly contagious and presents with general malaise, fever, and mucous patches. The disease remains latent in about one third of patients; one third of patients go into remission; and the final one third of patients have tertiary syphilis, which may occur years after the initial infection with a slowly progressive disease and may involve the central nervous system (neurosyphillis), the aorta, and gummas.
Patients can present with various symptoms involving the head and the neck and may have generalized lymphadenopathy. Syphilis may cause laryngitis, vocal fold paralysis, or dysphagia. It may also cause granulomatous involvement of the oral and nasal mucosa, septal perforation, and a chancre in the oral mucosa. Otologically, syphilis presents as an abrupt, fluctuating sensorineural hearing loss. Gummas may also be found in the temporal bone.
Diagnosis is aided by performing the specific fluorescent treponemal antibody absorption (FTA-ABS) test. Treatment consists of penicillin, with steroids reserved for otologic involvement.
For patient education information, see the Sexual Health Center, as well as Syphilis.
Fungal Granulomatous Diseases
Histoplasmosis
The fungus Histoplasma capsulatum causes histoplasmosis. It most commonly occurs in the Ohio and Mississippi River Valleys. The organism is spread by airborne transmission of avian droppings. Persons who are immunocompromised may present with painful ulcers on the lips, the gingiva, the tongue, the pharynx, or the larynx. Culture on Sabouraud medium, skin test, complement fixation, or latex agglutination can be used to establish a diagnosis. Patients are treated with amphotericin B.
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis. The common triad consists of cutaneous disease, pulmonary involvement, and constitutional symptoms. Individuals with blastomycosis often have either an immunodepressive condition or diabetes mellitus.
Establishing a diagnosis can be difficult. Blastomycosis is often mistaken for pneumonia sensu latu, malignant tumors, and tuberculosis. To confirm the diagnosis, sputum culture on Sabouraud medium is performed. New serologic tests include the sandwich enzyme immunoassay (sensitivity 88%, specificity 100%) and a 120kd antigen radioimmunoassay (sensitivity 85%, specificity 100%). Histopathologic examination reveals a pseudoepitheliomatous hyperplasia and a single bifringent, broad-based bud.
Rhinosporidiosis
Rhinosporidiosis is caused by the sporangium Rhinosporidium seeberi, which is primarily found in southern India. This lesion is spread by contaminated water. The physical examination may reveal strawberry lesions, which are painless, warty lesions found on the mucous membranes of the head and the neck, particularly in the nasal mucosa. Treatment consists of surgical excision and oral antifungal agents.
Congenital Granulomatous Diseases
Granulomatous diseases also occur due to hereditary defects in immunity. Patients with these diseases present with recurrent or chronic infections. Typically, defects in neutrophils and monocytes are the etiology of recurrent infections, but other deficiencies may contribute.
Chronic granulomatous disease is a rare disorder of phagocytic cells. It is a congenital disorder (66% X linked, 33% autosomal recessive) that results in a defect in the mechanism for intracellular killing of bacteria by neutrophils and macrophages. This translates clinically into patients with recurrent infections from catalase-positive organisms (ie, Staphylococcus aureus), gram-negative bacteria, and fungi. This will result in eczema, osteomyelitis, abscesses, and granulomas. The treatment is frequent courses of antibiotics and surgical debridement of infections. Treatment can also consist of interferon gamma, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) to stimulate neutrophils.
Chediak-Higashi syndrome is an autosomal recessive disorder that presents with oculocutaneous albinism, neuropathy, neutropenia, and possibly malignancy.
Job syndrome is an extremely rare disorder caused by extremely elevated levels of IgE and eosinophils. Patients often first present with eczema and characteristic staphylococcal abscesses. Prophylactic antistaphylococcal antibiotics are often used but have not been proven to be effective.
-
Mass on the right side of the neck in a patient with histiocytosis X.
-
Histology of eosinophilic granuloma.
-
Mass on the right side of the neck in a patient with atypical tuberculosis.





