Overview
Sarcomas are relatively uncommon tumors, accounting for 1% of all malignancies. Sarcomas are classified according to the histologic tissue from which they are derived, and more than 30 histologic subtypes have been described. Soft tissue sarcomas arise from the mesenchyme, including muscle, endothelial cells, cartilage, and supporting elements. This subclassification also includes tumors of peripheral nerve origin. Approximately 80% of sarcomas originate from soft tissues, while 20% arise from bone. Fewer than 5000 cases of sarcomas occur per year in the United States. [1, 2] Imaging studies are used to demonstrate the extent of tumor involvement. [3] Management depends on the type of sarcoma, although excision is the treatment of choice for many varieties. See the image below.
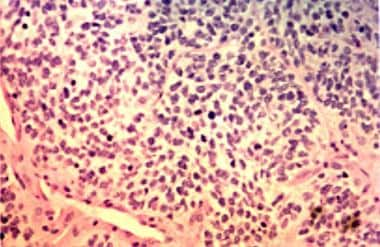 Photomicrograph (hematoxylin-eosin) shows rhabdomyosarcoma with hypocellular and hypercellular areas. Round cell and spindle cell components are present. Spindle cells are marked by their fusiform shape and eosinophilic cytoplasm.
Photomicrograph (hematoxylin-eosin) shows rhabdomyosarcoma with hypocellular and hypercellular areas. Round cell and spindle cell components are present. Spindle cells are marked by their fusiform shape and eosinophilic cytoplasm.
Approximately 15-20% of sarcomas occur within the head and neck; the paranasal sinuses and neck are the most frequent sites of origin. Unlike the development of squamous cell carcinoma, the development of sarcomas is unrelated to smoking and alcohol use. Approximately 80% of head and neck sarcomas occur in adults, and 10-20% occur in children. Certain sarcomas are related to genetic syndromes (eg, Li Fraumeni syndrome, osteosarcoma), environmental exposures (eg, radiation, multiple sarcoma types), and medical conditions (eg, lymphedema, angiosarcoma). When all anatomic sites are considered, the most common histologic type is malignant fibrous histiocytoma.
In the head and neck, the most common sarcoma in children is rhabdomyosarcoma; in adults, osteosarcoma, angiosarcoma, malignant fibrous histiocytoma, and fibrosarcoma are most common.
Most head and neck sarcomas occur with localized disease. Regional metastases occur in 10-15% of head and neck sarcomas overall; most of these arise from high-grade primary lesions. At presentation, distant metastases are rare in the absence of regional metastases, and the presence of nodal metastases should prompt a search for distant metastases. By far, the most common site of distant metastases is the lung, followed by the liver and bone. Local recurrence, which is the most common reason for treatment failure, is common.
Computed tomography (CT) scanning is useful in demonstrating bony involvement and tumor calcification. Contrast material infusion is usually required to fully appreciate the relationship of the tumor to adjacent vessels. Tumor margins can be difficult to distinguish from the fascia of adjacent musculature on CT scans, and resolution may be limited by artifacts in dense bone. Magnetic resonance imaging (MRI) is superior to CT scanning in its ability to depict the extent of soft tissue involvement by sarcomas. Intracranial extension is better delineated on MRI. The information obtained from both CT scanning and MRI is complementary, and both studies are often required in the evaluation of extensive lesions. Occasionally, imaging findings can suggest the correct diagnosis, but definitive diagnosis requires histologic evaluation of a representative biopsy specimen.
In a large study using the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program, Peng et al calculated that cause-specific survival rates in adults with sarcomas of the head and neck, at 2, 5, and 10 years, are 76%, 66%, and 61%, respectively, while these rates in children were calculated to be 84%, 73%, and 71%, respectively. In addition, multivariate analysis indicated that in adults, factors linked to a greater cause-specific survival rate include male gender, absence of radiation therapy, and stage I disease, although a propensity-matched model did not show an association between presence or absence of radiation therapy and cause-specific survival. The study employed data from 11,481 adult cases and 1244 pediatric cases. [4]
Sarcoma Classification
Sarcomas are commonly classified according to their site of origin: soft tissues or bone. Sarcomas involving cartilage (chondrosarcoma) and peripheral nerve tissue (malignant schwannoma) are included in the soft tissue category because they share a mesenchymal origin. Some soft tissue sarcomas may arise in bone (eg, fibrosarcoma, malignant fibrous histiocytoma), and sarcomas of bone may have extraosseous manifestations; therefore, the classification of soft tissue versus bone sarcoma depends more on histologic findings than clinical findings. [5]
Soft tissue sarcomas include the following (see Types of Soft Tissue Sarcomas):
-
Angiosarcoma
-
Hemangiopericytoma
-
Malignant fibrous histiocytoma
-
Synovial sarcoma
-
Chondrosarcoma
-
Rhabdomyosarcoma
-
Malignant schwannoma
-
Liposarcoma
-
Leiomyosarcoma
-
Fibrosarcoma
-
Alveolar soft part sarcoma (ASPS)
-
Kaposi sarcoma (KS)
Bone sarcomas include the following (see Bone Sarcomas):
-
Osteosarcoma
-
Ewing sarcoma
So-called radiation-induced sarcomas (RISs) are not a separate entity. They include a heterogeneous group of sarcomas that occur in tissue that has been irradiated. [6]
Staging of Soft Tissue Sarcomas
Two staging systems for soft tissue sarcomas are listed below. Modifications are made for selected tumor types (eg, synovial sarcoma, fibrous histiocytoma, rhabdomyosarcoma) and are described (see Types of Soft Tissue Sarcomas).
American Joint Committee on Cancer (AJCC) and International Union Against Cancer (UICC) staging system for soft tissue sarcomas
Primary tumor
Classifications include the following:
-
Tx - Primary tumor cannot be assessed
-
T0 - No evidence of primary tumor
-
T1 - Tumor less than 5 cm in greatest dimension (T1a, superficial; T1b, deep)
-
T2 - Tumor greater than 5 cm in greatest dimension (T2a, superficial; T2b, deep)
Regional lymph nodes
Classifications include the following:
-
Nx - Lymph nodes cannot be assessed
-
N0 - No lymph nodes metastases
-
N1 - Lymph nodes metastases present
Distant metastases
Classifications include the following:
-
Mx - Distant metastases cannot be assessed
-
M0 - No distant metastases
-
M1 - Distant metastases present
Histopathologic grade
Classifications include the following:
-
Gx - Grade cannot be assessed
-
G1 - Well-differentiated
-
G2 - Moderately differentiated
-
G3 - Poorly differentiated
-
G4 - Undifferentiated
Combined
Classifications include the following:
-
IA (G1-2, T1a-b, N0, M0) - Low-grade, small, and superficial or deep tumor
-
IB (G1-2, T2a, N0, M0) - Low-grade, large, and superficial tumor
-
IIA (G1-2, T2b, N0, M0) - Low-grade, large, and deep tumor
-
IIB (G3-4, T1a-b, N0, M0) - High-grade, small, and superficial or deep tumor
-
IIC (G3-4, T2a, N0, M0) - High-grade, large, and superficial tumor
-
III (G3-4, T2b, N0, M0) - High-grade, large, and deep tumor
-
IV (any G, any T, N1, M0) - Any metastasis
Memorial Sloan-Kettering staging system for soft tissue sarcomas
See the list below:
-
Size (cm) - Less than 5 cm is favorable; greater than 5 cm is unfavorable
-
Depth of invasion - Superficial is favorable; deep is unfavorable
-
Grade - Low is favorable; high is unfavorable
Stage grouping based on Memorial Sloan-Kettering staging system for soft tissue sarcomas
See the list below:
-
Stage 0 - 3 Favorable signs
-
Stage I - 2 Favorable signs
-
Stage II - 1 Unfavorable sign or 1 favorable sign and 2 unfavorable signs
-
Stage III - 3 Unfavorable signs
-
Stage IV - Distant metastases
Types of Soft Tissue Sarcomas
A study by Lee et al indicated that in head and neck soft tissue sarcoma, current size cutoff points of 2 and 4 cm are not prognostic for T1-T3 tumors. They reported, however, that outcome can be assessed via a 5 cm cutoff point for tumor stage. Employing recursive partitioning analysis (RPA), the investigators developed a stage grouping system, determining the following 5-year overall survival rates for head and neck soft tissue sarcoma [7] :
-
RPA Stage I (pT1-3N0-1G1-2M0) - 71.2%
-
RPA Stage II (pT4abN0-1G1-2M0/pT1-3N0-1G3-4M0) - 53.4%
-
RPA Stage III (pT4abN0-1G3-4M0) - 17.5%
Angiosarcoma
Angiosarcomas account for fewer than 1% of all sarcomas. This rare malignancy arises from endothelial cells of either lymphatic or vascular origin. When the cell of origin can be identified as lymphatic, the lesion is termed a lymphangiosarcoma; when vascular derivation can be ascertained, the correct term is hemangiosarcoma. Often, the cell of origin cannot be determined with certainty; hence, the more general term angiosarcoma is used.
The exact histogenesis of angiosarcomas remains unclear. Approximately half of these lesions occur in the head and neck. The scalp and face are the most common sites of involvement. Angiosarcomas typically occur in those aged 50-70 years, and males are more commonly affected than females, with a 3:1 ratio. While most patients are asymptomatic at the time of diagnosis, some have pain or bleeding at the tumor site. Conditions associated with angiosarcoma include long-standing lymphedema and prior irradiation. Exposure to vinyl chloride is associated with angiosarcomas of the liver, but this association has not been reported with head and neck angiosarcomas.
Trauma, either in the form of direct tissue injury or an adjacent foreign body or prosthesis, has been suggested as an etiologic factor; however, a causal relationship has not been proven. The AJCC staging system for soft tissue sarcomas is used to grade angiosarcomas. Low-grade lesions appear as raised, red or purple, painless papules or nodules. Often, small lesions surround a central lesion. High-grade lesions are usually multiple and ulcerated, with extensive local infiltration.
On microscopic examination, the most common histologic pattern of angiosarcomas involves abundant atypical endothelial cells that form irregularly arranged anastomotic sinusoids and dilated vascular spaces. Anastomoses between vascular channels are common, and endothelial cells with hyperchromatic nuclei line the vascular channels. A prominent lymphoid component may be present. Low-grade lesions have minimal cellular atypia and few mitotic figures, and 1 or more nodules may be present in the dermis. Proliferating endothelial cells project into vascular lumina.
High-grade lesions have greater pleomorphism, more hyperchromatic nuclei, and more mitotic figures. The number of mitotic figures has been suggested to have prognostic significance. Adjacent satellite lesions are common, and endothelial-lined papillae are present in the adjacent soft tissue beyond the central tumor; this finding suggests a multicentric origin, which may explain the high incidence of local recurrence after surgical excision.
Immunohistochemical staining for endothelial cell markers, including factor VIII, vimentin, CD31, CD34, ulex europeus, laminin, BNH9, and monoclonal antibody EN4 is used to diagnose angiosarcoma. Factor VIII staining may be variable or absent in angiosarcomas of lymphatic origin. Reticulin staining demonstrates the associated network of reticulin fibers associated with endothelial proliferation. Electron microscopy demonstrates Weibel-Palade bodies, which are microtubular bundles specific to tumors of vascular derivation.
Wide local excision is the mainstay of treatment. Prognosis appears to be most affected by the ability to perform complete surgical excision, as evidenced by the fact that local recurrence occurs in more than 50% of cases and is the most common pattern of treatment failure. Most recurrences are evident within 2 years of initial treatment. Regional metastases are less common, occurring in fewer than 20% of cases in most large series. Cervical lymph node metastases are more common in lesions arising from the scalp, and regional lymph node dissection is recommended in patients with scalp lesions or palpable lymphadenopathy.
Distant metastases occur in 30-50% of cases, with the lungs and liver most frequently involved. Five-year survival rates of 12-41% have been reported; most investigators report survival rates lower than 30%. A correlation between primary tumor size and survival appears to exist; lesions smaller than 7 cm are associated with a better prognosis. Adjunctive radiation therapy is used to improve locoregional control in high-grade tumors, tumors in which resection margins are close, and in scalp lesions. Doses of 6000-7000 rads (6000-7000 cGy) should be delivered to a field that extends several centimeters beyond the gross borders of the tumor because of the local multicentric nature of angiosarcomas. The benefit of adjuvant chemotherapy has not been established.
Work in molecular genetics holds promise in improving survival rates in patients with angiosarcomas. Current investigations seek to determine if an association between angiosarcoma histogenesis and mutations in the p53 tumor suppressor gene, the c-ras proto-oncogene, and the erbB proto-oncogene exist.
Hemangiopericytoma
Hemangiopericytomas are rare and account for fewer than 1% of vascular neoplasms. These tumors arise from the vascular pericytes of Zimmerman, which occur around capillaries and postcapillary venules. Hemangiopericytomas are distinct from the vascular angiosarcomas. Pericytes are mesenchymally derived and retain the capability of smooth-muscle differentiation. The biologic function of pericytes is not known, although a role in vascular capacitance is suggested. Approximately 15-30% of hemangiopericytomas occur in the head and neck; the sinonasal tract is the most common site.
No sex predilection exists, and 90% of hemangiopericytomas arise in persons aged 50-70 years. No etiologic factors are known. Rarely, hemangiopericytomas have been reported to be associated with a paraneoplastic syndrome; osteomalacia, hypoglycemia, and hypertension have been described. Approximately 10% of hemangiopericytomas occur in children; hemangiopericytomas tend to have a much more indolent course in this group.
At gross examination, hemangiopericytomas typically appear as bland polypoid growths in the sinonasal tract. In other regions, the typical manifestation is that of a slowly growing, painless mass. When they are large, a bruit may be auscultated. Biopsy can be associated with significant bleeding. Histologically, hemangiopericytomas consist of closely packed, small, uniform cells that are closely associated with capillaries and larger vascular channels. Dilated vascular channels have been described as having a staghorn appearance.
The tumor cells have scanty pink cytoplasm and hyperchromatic nuclei. Immunohistochemical findings are positive with vimentin and reticulin but negative with other endothelial cell markers. Significant mitotic activity is uncommon; however, the diagnosis of malignancy depends on the detection of mitotic bodies, which is predictive of clinical behavior and prognosis. Any mitotic activity is a sign of malignancy. Lesions associated with a poorer prognosis are those larger than 6.5 cm with more than 3 mitotic figures per 10 high-power fields, necrosis, and hemorrhage. Survival rates decrease dramatically when more than 4 mitotic figures per 10 high-power fields are present.
Complete surgical excision is the treatment of choice for patients with hemangiopericytomas. Compared with other sarcomas of the head and neck, hemangiopericytomas are associated with a better prognosis. Local recurrence rates of 10-50% have been reported; fewer than 20% of patients have local recurrence after surgical excision. Good local control rates have been attributed to an indolent course for this particular lesion as well as to its superficial location in the head and neck and typical polypoid presentation in the sinonasal tract; this location makes this lesion more amenable to complete surgical excision.
Regional and distant metastases occur in fewer than 10% of cases and are usually associated with recurrent disease at the primary site. An overall 10-year survival rate of 70% has been reported for hemangiopericytomas; however, the 10-year survival rate in patients with lesions with aggressive histologic characteristics (eg, > 4 mitoses per 10 high-power fields, necrosis, hemorrhage, large size) decreases to 29%.
Hemangiopericytomas are considered relatively resistant to radiation therapy; thus, radiotherapy is only an adjuvant therapy in incompletely excised lesions, tumors with high-grade histologic findings, or recurrent tumors. Chemotherapy is not standard in the management of hemangiopericytomas, although adjuvant chemotherapy may have a role in the management of distant metastatic disease and when primary resection is not possible without significant morbidity or disfigurement. Vincristine, doxorubicin, and cyclophosphamide delivered in the preoperative setting have been reported to significantly decrease in the size of the primary tumor. Anecdotal reports suggest that Adriamycin-based chemotherapy or alfa-interferon may be of benefit in patients with pulmonary metastases or unresectable primary tumors.
Malignant fibrous histiocytoma
Malignant fibrous histiocytoma is the most common soft tissue sarcoma in adults. Malignant fibrous histiocytomas are most common in the soft tissues of the abdomen and extremities, although 23% occur in osseous sites. Approximately 1-3% of malignant fibrous histiocytomas occur in the head and neck. Peak occurrence is in persons aged 50-70 years; the angiomatoid subtype occurs more frequently in young adults. A slight male predominance is observed. The most common sites of occurrence in the head and neck are the sinonasal tract, soft tissues of the neck, craniofacial bones, and salivary glands. The development of malignant fibrous histiocytoma is associated with previous radiation treatment and, less commonly, with the injection of silica as a sclerosing agent.
Malignant fibrous histiocytoma is thought to originate from fibroblasts or from a mesenchymal precursor cell that can differentiate into fibroblasts and histiocytes. Various histologic subtypes exist; these include storiform-pleomorphic, myxoid, xanthomatous or inflammatory, angiomatoid, and giant cell subtypes (see World Health Organization classification of fibrous histiocytomas). The storiform-pleomorphic variant is the most common subtype of malignant fibrous histiocytoma. This subtype has distinct populations of spindle cells, histiocytes, and giant cells; the spindle cells assume a storiform or cartwheel orientation. Areas of neutrophil infiltration and collagen production may be present.
The myxoid subtype is the next most common form, and it is distinguished by hypocellular myxoid areas with abundant mucopolysaccharide production, in conjunction with cellular components that are analogous to those in the storiform-pleomorphic or xanthomatous subtypes. Inflammatory or xanthomatous malignant fibrous histiocytoma is characterized by the proliferation of histiocytes, xanthomatous cells, and neutrophils, and it may be difficult to distinguish from an inflammatory disorder. Many patients are febrile and have peripheral granulocytosis. Angiomatoid malignant fibrous histiocytoma is characterized by sheets of histiocytes in association with blood-filled spaces.
Giant cell malignant fibrous histiocytoma involves multinucleated giant cells, histiocytes, and fibroblasts. Often, osteoids accumulate at the periphery of the lesion. The angiomatous and myxoid subtypes have the best prognosis because of a lower propensity for systemic metastases.
The World Health Organization classification of fibrous histiocytomas is as follows:
-
Benign
Fibrous histiocytoma
Dermatofibroma
Deep histiocytoma
Juvenile xanthogranuloma
Reticulohistiocytoma
Xanthoma
-
Intermediate
Atypical fibroxanthoma
Dermatofibrosarcoma protuberans
Giant cell fibroblastoma
Plexiform fibrohistiocytic tumor
Angiomatoid fibrous histiocytoma
-
Malignant
Storiform-pleomorphic tumor
Myxoid tumor
Giant cell tumor
Xanthoma (inflammatory tumor)
Immunohistochemistry is of little value in the diagnosis of malignant fibrous histiocytoma because no specific marker for these lesions exists. The diagnosis is made on the basis of the histologic appearance. Immunohistochemical staining can be used to differentiate malignant fibrous histiocytoma from other malignancies.
The overall mortality rate with malignant fibrous histiocytoma is 40-45%. Age appears to be correlated with mortality; one group reported a mortality rate of 72% in patients older than 60 years and a 33% mortality rate in patients aged 20-39 years. Local recurrence is present in 20-42% of cases; regional lymph node involvement occurs in 0-15% of cases; and distant metastases are found in 25-35% of cases. Recurrence usually occurs within 2 years of treatment. Recently, the chromosomal abnormality 19p+ has been demonstrated in malignant fibrous histiocytoma. Local recurrence is more common in tumors with 19p+. Distant metastases are more common in high-grade tumors and tumors larger than 5 cm.
Complete surgical resection is the treatment of choice for malignant fibrous histiocytoma. Because regional lymph node metastases are present in fewer than 15% of patients, lymph node dissection is not indicated in the absence of palpable cervical metastases, with the exception of tumors in the oral cavity, which appear to have a higher incidence of regional nodal involvement. Adjuvant radiotherapy is reserved for lesions with close or positive resection margins; systemic chemotherapy is used for distant metastatic disease.
Synovial sarcoma
Synovial sarcomas represent 6-10% of all soft tissue sarcomas. Only 3-10% of synovial sarcomas arise in the head and neck. These tumors occur in younger people; peak incidence is in those aged 20-40 years. Males are affected twice as often as females. The hypopharynx and retropharynx are the most common sites of involvement in the head and neck. The most common presentation is that of a painless mass; various associated symptoms may be present, depending on the location of the tumor. Calcifications are present in more than 50% of tumors and may be noted on radiography; this finding suggests the diagnosis. MRI demonstrates a characteristic nonmucosal mass that is homogeneous on T1-weighted images and heterogeneous on T2-weighted images.
Synovial sarcomas do not arise from synovial tissue; rather, they originate from pluripotential mesenchymal cells, and they rarely occur within joint spaces. The gross pathologic appearance is that of a white matter or gray matter mass, with a consistency that varies from firm, calcified, or fibrous to soft, cystic, or mucoid. Mesenchymal cells differentiate into 2 components: an epithelial-like cell layer and a connective-tissue layer of spindle-shaped cells.
Three subtypes of synovial sarcoma are described: biphasic, monophasic, and poorly differentiated. Biphasic synovial sarcomas are composed of epithelioid and spindle cells. Usually, the spindle cell component predominates. Mast cells, mitoses, areas of calcification, and scant collagen production are typical of biphasic synovial sarcoma. The epithelioid cells form pseudoglandular cavities filled with mucin, which stains positively with Alcian blue, mucicarmine, and periodic acid-Schiff (PAS) stains. Mesenchymal mucin is associated with the spindle cell component and stains positively with Alcian blue.
Monophasic synovial sarcoma is composed of 1 cellular type and may be derived from epithelioid or spindle cells. Both epithelioid and spindle cells stain positively for cytokeratin and epithelial membrane antigen (EMA). Spindle cells stain positively with vimentin, a mesenchymal marker, whereas epithelioid cells stain negatively with vimentin. A rare poorly differentiated subtype has been described. These tumors may consist predominantly of epithelioid cells, spindle cells, or a small cell variant that forms rosettes.
A reciprocal translocation, t(X;18)(p11.2;q11.2), has been identified in monophasic and biphasic synovial sarcoma. The presence of this translocation confirms the diagnosis of synovial sarcoma. Chromosome 18 contains the SYT gene, which fuses with SSX1 or SSX2 from chromosome X. The SYT-SSX1 fusion is associated with biphasic variants; SYT-SSX2 fusion is associated with monophasic variants.
The AJCC classifies all synovial sarcomas as high grade. Regional lymph node metastases occur in fewer than 5% of synovial sarcomas, whereas distant metastases, most frequently to the lung, occur in 50% of cases. Fewer than 20% of patients with metastatic disease live longer than 2 years after diagnosis. Isolated pulmonary metastases may be amenable to thoracotomy, with improved survival in 20-40% of patients when resection is feasible. Because of the rarity of cervical metastases, neck dissection is not indicated in the absence of palpable metastatic disease. Local recurrence is present in approximately 40% of patients.
Factors associated with a poor prognosis include increasing age, tumor size greater than 5 cm, and mitotic activity. Overall 5-year survival rates for synovial sarcoma of the head and neck are 36-51%, with a 10-year survival rate of 11.2-30%. Survival rates in children are 54-65% at 5 years.
A retrospective, single-institution study by Gopalakrishnan et al similarly indicated that in head and neck synovial sarcoma, a poorer progression-free survival (PFS) is predicted by a tumor size of 5 cm or greater, as well as by a primary presentation in the neck’s soft tissues. The investigators found that the median PFS for all 44 of the study’s patients was 4.6 years, with local or distant recurrence developing in 20 patients (45%). [8]
Surgical excision combined with postoperative radiation therapy is the primary treatment for synovial sarcoma. Local recurrence rates are 60-90% when surgery is the sole therapy. The addition of adjuvant radiotherapy to surgery reduces the recurrence rates to 28-49%. Doses of at least 65 Gy must be used for any survival advantage. Chemotherapy with ifosfamide compounds appears to be of benefit in the treatment of distant metastases.
Chondrosarcoma
Chondrosarcomas arise in cartilage or bone and account for 20% of primary malignant bone tumors. Approximately 5-10% of chondrosarcomas are located in the head and neck; they most commonly involve the larynx, followed by the maxilla, mandible, and base of the skull. Chondrosarcomas are the most common type of laryngeal sarcoma and the second most common type of sarcoma arising from bone in the head and neck. Chondrosarcomas occur in persons of many ages, but incidence peaks in those aged 30-50 years. Laryngeal lesions are more prevalent in males, but no sex predilection exists with other sites.
Symptoms arise from the involvement of adjacent structures. Approximately 80% of chondrosarcomas are primary lesions arising in healthy tissue, with no know etiologic factor. Secondary chondrosarcomas, which arise from preexisting lesions, have been noted in association with exostosis; Paget disease; fibrous dysplasia; Maffucci syndrome; and exposure to irradiation, lucite, beryllium, zirconium, and thorium dioxide. Extraosseous tumors have been described and are believed to arise from cartilaginous differentiation of primitive mesenchymal cells.
Chondrosarcomas are slow growing, locally invasive lesions, and symptoms are referable to the site of origin. Calcification is common and may be helpful in making a diagnosis using radiographs. On CT scans, laryngeal chondrosarcomas appear to be confluent with the laryngeal cartilages, most often the cricoid cartilage. In other sites, chondrosarcomas appear osteolytic with sun-ray spiculation of bone within the lesion. The distinction between a benign chondroma and chondrosarcoma can be made with radiologic findings on the basis of tumor size; chondromas are smaller than 3 cm, whereas chondrosarcomas are 3 cm or larger.
On gross examination, chondrosarcomas are firm, lobular, white or bluish submucosal lesions that are larger than 2 cm. A myxoid variant with a more gelatinous consistency has been described. Chondrosarcomas are highly cellular on microscopic examination, and they frequently have lacunae that contain multiple pleomorphic nuclei in a matrix of hyaline cartilage. Chondrosarcoma tumor cells form chondroids, but not osteoids; this feature distinguishes these lesions from osteosarcomas. Chondromas lack binucleated nuclei, they are less cellular than chondrosarcomas, and they are rarely larger than 2 cm.
Histologic grading of chondrosarcomas can be used to separate these lesions into well-differentiated (grade I), moderately differentiated (grade II), and poorly differentiated (grade III) lesions. Well-differentiated lesions contain small, dark nuclei and scant-to-absent mitosis. The tumor matrix is variable, consisting of both chondroid and myxoid components that resemble hyaline cartilage; calcification is common. Moderately differentiated lesions have larger nuclei, greater cellularity, a matrix with a more predominant myxoid component, and occasional mitotic bodies (< 2 per 10 high-power fields).
Poorly differentiated lesions have even greater cellularity, with prominent nucleoli; mitotic bodies are common (2 or more per 10 high-power fields), and the matrix may contain fusiform spindled cells. A hemangiopericytic vascular pattern has been described in some poorly differentiated tumors. Most chondrosarcomas are well-differentiated grade I lesions; grade II and III lesions are associated with a poorer prognosis. High-grade lesions may contain areas of dedifferentiation that microscopically resemble other mesenchymal tumors, such as fibrosarcoma. Diagnosis depends on the identification of a lower-grade component that produces chondroid.
Surgical resection is the main treatment for chondrosarcomas, irrespective of site of origin. Regional and distant metastases are present in fewer than 10% of chondrosarcomas at initial diagnosis. Because of the low incidence of regional nodal involvement, neck dissection is not indicated in the absence of palpable adenopathy. Local recurrence is the most common cause of treatment failure and is present in 50% of patients. Adequacy of surgical resection is the main determinant of recurrence. Prognosis is dependent on the site of origin and tumor grade.
Chondrosarcomas arising in the larynx are associated with a better prognosis, although total laryngectomy is often required for complete removal. Conservation surgery is associated with a higher incidence of local recurrence. Chondrosarcomas arising in the nasopharynx or sinonasal tract are associated with a poorer prognosis. Grade III and dedifferentiated lesions are highly aggressive lesions that are associated with a higher incidence of local recurrence and distant metastases. Distant metastases are 3 times more likely in higher-grade lesions than in lower-grade lesions.
Chondrosarcomas are considered to be resistant to radiotherapy, and, in general, adjuvant radiation therapy is not used. However, survival rates increase with chondrosarcomas of the base of the skull with postoperative irradiation; this finding suggests that some lesions may be sensitive to radiation therapy. Chemotherapy does not have a demonstrable benefit in the management of chondrosarcoma, but chemotherapy is sometimes used for high-grade tumors with distant metastasis. Survival rates exceed 80% at 5 years and 70% at 10 years. High-grade lesions and distant metastases are associated with lower survival rates, which approach 50%. Metastases can develop years after initial presentation, and follow-up for much longer than 5 years is required.
Rhabdomyosarcoma
Rhabdomyosarcoma accounts for 19% of all sarcomas and is the most common soft tissue sarcoma in children. The head and neck are the most common sites of occurrence; 45% of rhabdomyosarcomas occur here. Rhabdomyosarcoma is primarily a pediatric disease; 80% of rhabdomyosarcomas occur in children younger than 12 years, and rhabdomyosarcoma is the seventh most common pediatric malignancy. A slight male predominance (1.5:1) exists, and the prevalence is increased in whites compared with prevalence in other races. The most commonly involved sites are the orbit, nasopharynx, temporal bone, and sinonasal tract. [9, 10]
Rhabdomyosarcoma is a malignant tumor of the skeletal muscle cells, and it arises from the rhabdomyoblast. The gross appearance varies according to the location. Nasopharyngeal tumors are usually tan or white, and they have a well-circumscribed polypoid or multinodular appearance. They may become large before diagnosis. Tumors arising from the ear or sinonasal tract tend to be smaller and usually appear as an aural or nasal polyp. A botryoid, or grapelike, multinodular appearance, termed sarcoma botryoides, has been described in sinonasal and nasopharyngeal locations. Orbital tumors typically appear with unilateral proptosis and a lid mass.
In the head and neck, four subtypes of rhabdomyosarcoma have been described: embryonal, alveolar, pleomorphic, and mixed. The embryonal subtype is the most common (>70%), and it typically involves round cells that resemble lymphocytes and spindled cells that contain elongated nuclei and eosinophilic cytoplasm. The degree of cellularity varies within the tumor, and a myxoid stroma is often present in areas of relative hypocellularity. Alveolar rhabdomyosarcoma involves areas of spaces (alveoli) lined by noncohesive round or oval cells. Solid areas are interspersed, and fibrous tissue may be present; multinucleated giant cells are common.
Pleomorphic rhabdomyosarcoma is the least common subtype and is more common in older patients. Large pleomorphic rhabdomyoblasts are characteristic findings, and they may be rounded with peripheral nuclei or strap-shaped with multiple nuclei arranged in a row. Mixed rhabdomyosarcoma involves more than one histologic subtype. Mitosis is common in all subtypes.
Rhabdomyosarcomas are immunoreactive to desmin and myoglobulin. Additionally, reactivity to muscle-specific actin occurs in more than 75% of cases. Tumor cells contain glycogen; therefore, they stain positively with the PAS reagent; myofibrils stain positively with phosphotungstic acid.
The outcome varies with the location of the primary tumor, tumor size, patient age, local recurrence, and metastasis (see UICC tumor, node, metastasis staging system for rhabdomyosarcoma). A study by Amer et al employing information from the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database found that among the various subtypes of rhabdomyosarcoma, the worst 5-year survival rates were associated with pleomorphic and alveolar lesions (26.6% and 28.9%, respectively). At 73.9%, embryonal rhabdomyosarcoma had the highest 5-year survival rate. [11]
The Intergroup Rhabdomyosarcoma Study Group has classified lesions as orbital and parameningeal (includes temporal bone and ear, paranasal sinuses, nasopharynx, and infratemporal fossa) and in association with all other head and neck sites. With this classification, 5-year survival rates exceed 90% in orbital tumors, 69% in parameningeal sites, and 80% in all other head and neck sites.
Multimodality therapy with combination chemotherapy (vincristine, actinomycin D, cyclophosphamide, Adriamycin) with external-beam radiation therapy and nonradical surgery is superior to any single-modality therapy. Surgery has a smaller role in the treatment of this sarcoma; surgery is performed for biopsy and tumor debulking.
A study by Vaarwerk et al indicated that salvage treatment with AMORE (Ablative surgery, MOulage technique brachytherapy, and REconstruction) can prove effective in patients with relapsed head and neck rhabdomyosarcoma, including those who previously underwent external-beam radiation therapy. The overall 5-year survival rate in the study’s salvage patients, all of whom had undergone prior external-beam radiation treatment, was 54%. Survivors at median 8.6-year follow-up included 3 of 9 patients (33%) with parameningeal head and neck rhabdomyosarcoma, 6 of 7 patients (86%) with orbital rhabdomyosarcoma, and neither of two patients with nonparameningeal head and neck rhabdomyosarcoma. [12]
A clinical staging system for rhabdomyosarcoma developed by the Intergroup Rhabdomyosarcoma Study Group is listed below (see Intergroup Rhabdomyosarcoma Study Group clinical staging system for rhabdomyosarcoma). With this staging classification, the 5-year survival rates are 83% for stage I disease, 70% for stage II disease, 52% for stage III disease, and 20% for stage IV disease.
The UICC tumor, node, metastasis staging system for rhabdomyosarcoma is as follows:
-
Primary tumor
T1 - Tumor confined to organ or tissue of origin
T2 - Tumor involving one or more contiguous organs or tissues
-
Regional lymph node
Nx - Lymph nodes cannot be assessed
N0 - No lymph nodes metastases
N1 - Lymph nodes metastases present
-
Distant metastases
Mx - Distant metastases cannot be assessed
M0 - No distant metastases
M1 - Distant metastases present
-
Histopathologic grade
Gx - Grade cannot be assessed
G1 - Well differentiated
G2 - Moderately differentiated
G3 - Poorly differentiated
G4 - Undifferentiated
-
Combined
Stage I - T1, Nx or N0, M0
Stage II - T2, Nx or N0, M0
Stage III - Any T, N1, M0
Stage IV - Any T, any N, M1
The Intergroup Rhabdomyosarcoma Study Group clinical staging system for rhabdomyosarcoma is as follows:
-
Group I - Localized disease with tumor completely resected and regional nodes not affected, with the following: (1) confinement to the muscle or organ of origin or (2) contiguous involvement (ie, infiltration outside the muscle or organ of origin), as through fascial planes
-
Group II - Localized disease with microscopic residual disease or regional disease and no residual or microscopic residual disease and the following: (1) grossly resected tumor with microscopic residual disease (nodes negative), (2) completely resected regional tumor (nodes positive or negative), or (3) regional disease with grossly resected involved nodes and evidence of microscopic residual disease
-
Group III - Incomplete resection or biopsy sample with gross residual disease
-
Group IV - Metastatic disease present at onset
Malignant schwannoma
Malignant schwannomas, also known as malignant peripheral nerve sheath tumors or neurofibrosarcomas, arise from peripheral or cranial nerves and account for 5-10% of all sarcomas. The lower extremity is the most commonly involved site; however, as many as 20% of malignant schwannomas occur in the head and neck. Malignant schwannomas may arise sporadically or in association with von Recklinghausen disease or neurofibromatosis type I (NF-1). The sporadic form most commonly arises in persons aged 40-60 years, and females are affected more often than males. Tumors arising in association with NF-1 most commonly occur in those aged 20-40 years, and males are affected more often than females. The malignant triton tumor is a rare lesion consisting of a peripheral nerve sheath tumor that contains foci of rhabdomyosarcoma. One third of triton tumors arise in the head and neck, and one third are associated with NF-1. An association with previous irradiation also exists.
The most common site of origin in the head and neck is the neck, followed by the nasal cavity and paranasal sinuses, nasopharynx, oral cavity, orbit, cranial nerves, and larynx. Symptoms vary according to the site involved, but the most common presenting symptom is a painful, enlarging mass. Paresthesias, weakness, and muscle atrophy may also be present. As many as 50% of patients with malignant schwannomas have evidence of NF-1 or a positive family history. About 5-15% of patients with NF-1 have malignant schwannomas. Therefore, a diagnosis of malignant schwannoma should suggest the possibility of NF-1, and a rapidly enlarging, painful mass in a patient with NF-1 should be suspected of harboring a malignant schwannoma.
At gross examination, a malignant schwannoma appears as a fusiform or nodular mass that is firm or fleshy, and its origin may clearly be peripheral nerve tissue. The term malignant schwannoma is misleading because tumors can arise from any nerve sheath cell, including perineural fibroblasts and fibroblasts, as well as Schwann cells. Microscopically, malignant cells appear spindled, contain scant cytoplasm, and are oriented in fascicles that imitate a herringbone pattern. Cellular pleomorphism is common.
Nuclei are hyperchromatic and have variable pleomorphism. In classic malignant schwannoma, nuclei have an elongated, wavy, buckled appearance. One or more mitotic bodies per high-power field are common. Foci of epithelioid cells may be present; these can confound diagnosis. Cartilage and bone may also be present in small amounts. Foci of rhabdomyosarcomas may be present and result in a rare malignant triton tumor. Grading of malignant schwannoma is based on the degree of cellular pleomorphism, mitotic activity, and cellularity. Most malignant schwannomas (80%) are high-grade lesions.
One half to two thirds of malignant schwannomas stain positively for S-100; myelin basic protein and Leu-7 are also expressed in most lesions. The extent of expression depends on the degree of Schwann cell differentiation. Staining with HMB 45 and desmin demonstrates negative results. Electron microscopy may reveal ultrastructural features of Schwann cells that aid diagnosis.
Management of malignant schwannoma is primarily surgical; adjuvant radiation therapy is often used because most of these tumors are high-grade. Cervical lymph node metastases are rare; however, distant metastases occur in 33% of cases and most commonly involve the lung or bone. Local recurrence is present in 50% of patients. The overall survival rate is 50%. Tumors smaller than 5.0 cm are associated with a more favorable prognosis; multiple tumors in association with NF-1 are associated with a poorer prognosis. The outcome appears to differ with the clinical setting in which the tumors arise. In sporadic malignant schwannoma, reported 5-year survival rates are 50-75%, whereas survival rates for malignant schwannoma associated with NF-1 are 15-30%. Chemotherapy may have a role in the treatment of inoperable disease, recurrent disease, or disease that persists despite initial therapy.
Liposarcoma
Liposarcomas are among the most common soft tissue sarcomas, second only to malignant fibrous histiocytoma in incidence, and they account for 16-18% of all soft tissue sarcomas. However, only 3-6% of liposarcomas occur in the head and neck. The incidence of liposarcomas peaks in those aged 30-60 years, and a slight male preponderance is observed. The most common sites involved in the head and neck are the larynx, neck, and cheek. Liposarcomas are extremely rare in children. Typically, liposarcomas are slow growing, painless masses. With the exception of lesions in the larynx, which can cause dysphagia and airway symptoms, most patients with liposarcomas are asymptomatic at presentation. [13]
Although the literature contains examples of liposarcomas that arise from preexisting lipomas, most authors believe that liposarcomas are distinct lesions that arise from primitive mesenchymal cells. The gross and microscopic appearances vary considerably and range from the yellow-tan lipomalike appearance with abundant adipose cells to a gray, gelatinous, myxoid appearance.
At gross examination, liposarcomas appear encapsulated, but at microscopic evaluation, infiltrating borders are common. Malignant lipoblasts occur with varying frequency among the histologic subtypes of liposarcoma, and they may be univacuolated (ie, signet ring type) or multivacuolated. Vacuoles result from fat deposition, which moves the nucleus to a lateral position in univacuolated cells and centers the nucleus in multivacuolated cells. Five subtypes of liposarcoma have been identified: well-differentiated, myxoid, round cell, pleomorphic, and dedifferentiated.
Well-differentiated liposarcoma, also known as lipomalike liposarcoma or atypical lipoma, occurs in 20-30% of cases. This subtype has foci of lipoblasts within a lesion that otherwise resembles a lipoma. Myxoid liposarcoma is the most common subtype and accounts for 30-50% of cases. This subtype is characterized by a hyaluronic acid-rich stroma with widely separated monomorphic cells that are fusiform or stellate in appearance. Lipoblasts are present in various stages of development.
Myxoid liposarcomas can be distinguished from soft tissue myxomas by the presence of a plexiform capillary network. Round cell liposarcomas account for approximately 15% of liposarcomas and are characterized by densely packed small round cells with little fat vacuolization and sparse stroma. These are thought to represent a poorly differentiated variant of the myxoid subtype. This developmental association is supported by the finding that the translocation t(12;16)(q13;p11) is present in both myxoid and round cell liposarcomas but is absent in other subtypes.
Pleomorphic liposarcoma occurs in 10-35% of cases and features bizarre multinucleated lipoblasts with extreme pleomorphism and disordered growth patterns. Dedifferentiated liposarcomas have been described. At histologic analysis, they have the appearance of a well-differentiated liposarcoma with areas of dedifferentiation with a nonlipoblastic sarcomatous component. These account for approximately 5% of all liposarcomas. In all liposarcomas, the degree of tumor differentiation is well-correlated with tumor behavior and prognosis. The well-differentiated and myxoid types are associated with a more benign course, with fewer cases of local recurrence and distant metastases than lipoblastic and pleomorphic subtypes. More than 1 subtype can be present within a single tumor.
Wide local excision is the treatment of choice for liposarcomas; wide margins are especially important for local control, given the degree of infiltration associated with these tumors despite their apparent capsules. Regional nodal metastases are rare. Distant metastases occur via hematogenous spread in 30% of patients, and they are associated with the more poorly differentiated round cell and pleomorphic subtypes. Fewer than 20% of myxoid tumors and 6% of well-differentiated tumors are associated with distant metastases.
Survival and local control is associated with the tumoral subtype. The 5-year survival rates for liposarcomas are 85-100% for well-differentiated liposarcoma, 77-95% for myxoid liposarcomas, 21-45% for pleomorphic liposarcomas, and 13-55% for round cell liposarcomas. The incidences of local recurrence are 29-62% for well-differentiated liposarcoma, 33-57% for myxoid liposarcoma, 43-73% for pleomorphic liposarcoma, and 63-86% for round cell liposarcoma.
The role of radiation therapy in the treatment of liposarcomas is a subject of continued debate, although liposarcomas, particularly the well-differentiated and myxoid subtypes, are sensitive to radiation therapy. Radiotherapy is suggested as an alternative to surgery in lesions in which surgical resection would cause significant and unacceptable cosmetic or functional defect and in the postoperative setting after incomplete tumor resection. Currently, chemotherapy appears to have a limited role in the treatment of liposarcomas, although doxorubicin- and dacarbazine-based chemotherapeutic regimens have been reported to confer a survival advantage when they are used in the treatment of advanced myxoid liposarcoma.
Leiomyosarcoma
Leiomyosarcomas are uncommon and account for 6% of all sarcomas. Only 3-4% of these occur in the head and neck region. Leiomyosarcomas arise from smooth muscle cells, which, in the head and neck, are located in blood vessel walls and erector pili muscles. The most common sites of occurrence in the head and neck are the oral cavity, nasal cavity and paranasal sinuses, skin, cervical esophagus, and larynx. No sex predilection exists; leiomyosarcomas are most common in those aged 50-60 years, but they can occur in persons from many age groups. An association between tumors of sinonasal origin and prior cyclophosphamide chemotherapy and radiation therapy has been suggested.
On gross examination, leiomyosarcomas appear smooth and well circumscribed, and they are unencapsulated and gray, tan, or pink. They may be polypoid or sessile. Microscopically, leiomyosarcomas are composed of fascicular bundles of spindle-shaped cells with centrally cigar-shaped or blunt nuclei and intensely eosinophilic cytoplasm. Multinucleated giant cells and nuclear palisades are common. Reticulin fibers are present, although they are characteristically absent in large areas of tumor. Some pathologists divide leiomyosarcomas into epithelioid or vascular variants, depending on the degree of epithelioid or vascular cells present within the smooth muscle sarcoma. The degree of mitotic activity and tumor size appear to be the most important predictors of malignant behavior. One or more mitotic bodies per 5 high-power fields and a tumor larger than 2.5 cm are indications of aggressive behavior.
Immunohistochemical stains aid in diagnosing leiomyosarcoma. Leiomyosarcomas characteristically stain positive for smooth muscle–specific actin and vimentin. Reticulin stains also have positive results, depending on the degree of reticulin present. Staining for desmin has variable results, with an inverse relationship to the vascularity of the tumor. Some leiomyosarcomas may express cytokeratin, the presence of which can confound accurate diagnosis.
Leiomyosarcomas are locally aggressive neoplasms, and local recurrence is common. Metastases occur via hematogenous spread and most commonly involve the lungs. As a result, cervical metastases occur in fewer than 15% of patients and usually result from contiguous spread of the primary tumor. Cervical metastases typically occur late in the course of the disease, as opposed to distant metastases, which are present in 20% of patients at presentation. The average reported duration of survival is 48 months; patients with cutaneous lesions have a better prognosis than patients with lesions on other sites.
Wide local excision is the treatment of choice for primary and recurrent disease. Prognosis appears to be related to the site and extent of the primary tumor. Lesions arising from the skin, nasal cavity, and larynx are associated with a better prognosis than lesions in other sites in the head and neck, probably because these sites are more amenable to complete surgical resection. Combination chemotherapy and radiation therapy are used as adjuncts, but they do not appear to affect disease progression. Leiomyosarcoma is often poorly differentiated, particularly in patients who receive radiotherapy as treatment (alone or adjuvant). Surgical treatment has shown to increase the odds of a patient’s survival. [14]
Fibrosarcoma
Fibrosarcomas are relatively uncommon tumors and account for 12-19% of soft tissue sarcomas. More than half of all tumors arise in the lower extremities; approximately 10% occur in the head and neck, most commonly in the sinonasal tract and neck. Fibrosarcomas arise from fibroblasts and may be confused with other tumors with a prominent spindle cell component and collagen production; these include malignant fibrous histiocytoma, spindle cell carcinoma, malignant schwannoma, and synovial sarcoma. Between one third and one half of cases previously classified as fibrosarcoma have been reclassified as other lesions because of improved recognition of other histologically similar entities.
The development of fibrosarcoma is associated with previous radiation therapy or burn injury; tumors are reported to arise in irradiated sites or burn scars. The average time of onset of fibrosarcoma after tissue injury is 10 years after irradiation and more than 30 years after burn injury.
Fibrosarcomas may arise in patients of any age; a slight male predominance exists. Most cases occur in those aged 30-60 years. An infantile variant that occurs in patients younger than 5 years appears to represent a distinct subtype and is associated with a better prognosis. Clinically, fibrosarcomas most commonly manifest as painless, gradually enlarging masses; symptoms associated with increasing size vary according to the location of the tumor. Fibrosarcomas are homogeneous and nonenhancing on CT scanning, and they may cause bone remodeling. On T1- and T2- weighted MRI, fibrosarcoma has low or intermediate signal intensity.
At gross examination, fibrosarcomas are well-circumscribed, apparently encapsulated, firm, tan-gray lesions, but microscopic invasion is the rule rather than the exception. Fibrosarcomas are divided into well-differentiated and poorly differentiated subtypes based on the degree of cellular uniformity, collagen production, and mitotic bodies. Well-differentiated or low-grade tumors have a uniform spindle-cell appearance, eosinophilic cytoplasm, tapered nuclei arranged in an interlocking fascicular or herringbone pattern, and substantial collagen production. Poorly differentiated or high-grade lesions have greater cellular variability, with hyperchromatism, an increased number of mitotic figures, scant collagen production, and a greater degree of necrosis and hemorrhage. The infantile variant resembles the adult variant, and although no inheritance pattern has been described, an association with trisomy of chromosomes 8, 11, 17, and 20 has been reported.
Fibrosarcoma must be differentiated from malignant fibrous histiocytoma, fibromatosis, leiomyosarcoma, malignant schwannoma, melanoma, spindle cell carcinoma, and nodular fasciitis. Fibrosarcomas are largely unreactive at immunohistochemical staining; this feature can be useful in diagnosis. Immunohistochemical stains, such as S-100 or HMB-45, can be used to distinguish fibrosarcoma from melanoma or malignant schwannoma, and flow cytometry may be useful in differentiating fibrosarcoma from nodular fasciitis. Also, differences in histologic appearances are used to differentiate among tumor types.
Treatment consists of complete surgical excision, and postoperative radiation therapy is reserved for lesions with close or positive margins. High tumor grade; size larger than 5 cm; positive margins; and invasion of bone, skin, or neurovascular tissue are associated with unfavorable outcomes. Local recurrence occurs in 50-75% of cases, and this is the most common cause of death. Metastasis occurs via hematogenous spread. Distant metastasis occurs in 20-40% of cases and most often involves the lungs. Cervical metastasis occurs in 10% of cases and is believed to result from direct tumor extension; neck dissection is not indicated in the absence of palpable adenopathy. Five-year survival rates are 50-70%, and most recurrences are apparent within 2 years of initial treatment.
The infantile variant, which occurs in children younger than 5 years, has a much better prognosis than its adult counterpart. In children, 5-year survival rates are 85-95%. Factors associated with negative outcomes in adults do not appear to be correlated with negative outcomes in the infantile variant. Local recurrence is present in fewer than 30% of cases, and distant metastases occur in approximately 5% of cases; however, distant metastases have been reported to occur as long as 20 years after initial therapy. Children older than 10 years have survival rates similar to those of adults.
Alveolar soft part sarcoma
ASPS is a rare tumor, accounting for fewer than 1% of sarcomas. Head and neck involvement occurs in 27% of cases. The most common sites for ASPS in the head and neck are the orbit and tongue. Although this lesion can occur in patients of any age, most patients are younger than 40 years. The female-to-male ratio is 2:1. Tumors gradually enlarge and are painless; symptoms that occur are referable to the site involved. ASPS may initially manifest with symptoms resulting from cerebral metastases. ASPS tends to be highly vascular, and a bruit may be auscultated on examination. Both T1- and T2-weighted MRI shows high signal intensity.
Gross examination reveals a poorly circumscribed, friable, yellow or red lesion. Proliferation of blood vessels at the periphery of the lesion is common. Vascular and lymphatic infiltration can be identified at the margins of the tumor. The name ASPS is derived from its characteristic appearance at light microscopy, which is described as groups of epithelioid tumor cells in a highly vascular matrix. Grouped polygonal tumor cells with granular eosinophilic cytoplasm are arranged in an organoid configuration and separated by thin fibrovascular septa. Central areas within these nests of cells become necrotic, and the loss of architecture produces an alveolar appearance. Mitotic bodies are uncommon. Rhomboid and rod-shaped crystals are arranged in a sheaflike orientation in the cytoplasm of 75% of tumors. These produce positive results with PAS staining, are diastase resistant, and, when present, are diagnostic of ASPS.
Debate regarding the derivation of ASPS exists. ASPS is not related to any benign lesion. Some authors suggest a neuroendocrine origin, citing evidence of myelinated axon formation within the lesion. Others have not demonstrated this finding. Additionally, ASPS lacks the peripheral nerve myelin proteins P2 and P0, renin, and catecholamines. Most pathologists support the idea of a myogenous origin for ASPS because of the presence of MyoD1, myogen, and desmin in many lesions. Immunohistochemical stains for desmin are positive in 50% of tumors; reactivity to actin, MyoD1, vimentin, neuron-specific enolase, and S-100 is more variable. Mutation at the 17q25 site has been reported in ASPS, although the significance of this finding is unclear.
ASPS is associated with a deceptively indolent course. Distant metastasis is present in 25% of patients at initial presentation and most commonly involves the lung, bone, and brain. Cervical lymph node involvement is uncommon and occurs in 7% of cases. Surgical excision is the treatment of choice; elective neck dissection is not indicated because of the low incidence of cervical metastases. However, local recurrence and metastatic involvement can occur over a long period, contrary to other head and neck sarcomas. Local recurrence occurs in 20% of patients. Approximately 60% of patients who are free of metastases at initial presentation have distant metastases at some point after initial treatment. The survival rate at 5 years is reported to be 65%; however, when patients are monitored over time, the survival rate decreases to 38% at 10 years and 15% at 20 years.
Orbital lesions are associated with a disease-free survival period longer than that of other sites in the head and neck. Children have a better overall survival rate compared with that of adults. Adjuvant radiation therapy or chemotherapy has not been shown to provide any improvement in disease control or survival.
Kaposi sarcoma
In recent years, KS has received an increasing amount of attention because of its association with AIDS. AIDS-associated KS appears to behave differently than the better-known classic form.
Classic KS characteristically occurs in Mediterranean or Ashkenazi Jewish men aged 50-70 years and accounts for fewer than 1% of all neoplasms. Classic KS is an indolent neoplasm that is nonaggressive, and it often involutes spontaneously. Classic KS is not associated with HIV infection. The lower extremities, followed by the upper extremities, are most commonly affected sites. Only 10% of cases affect the head and neck, with most of these arising from cutaneous sites. Visceral involvement is uncommon and usually associated with concurrent cutaneous involvement. Classic KS is associated with other malignancies such as lymphoma, leukemia, and autoimmune disorders.
Nonclassic or epidemic KS is aggressive and occurs in immunocompromised patients, such as organ transplant recipients and patients with AIDS. KS occurs in fewer than 1% of transplant recipients, and the percentage of patients of Mediterranean or Jewish descent is disproportionately large; this finding suggests a genetic predisposition. In contrast, in the past, KS developed in as many as 40% of patients with AIDS, and KS was the presenting issue in 14% of patients in whom AIDS was previously undiagnosed.
Today, the prevalence of KS in patients with AIDS has declined to approximately 15% with the advent of highly effective antiretroviral therapy. Whether this decrease results from direct antiviral effects or immunologic restoration is unclear. In patients with AIDS, the head and neck are involved in more than 50% of cases; most often, the skin of the head and neck is involved, followed by mucosal surfaces, most frequently the palate.
On gross examination, KS appears as a painless, nonblanching, red to bluish macule or papule. Lesions are often multiple and may coalesce to form plaques that may become nodular or ulcerating. Histologically, the spindle cell is the dominant cell type, with variable pleomorphism and scant cytoplasm. The spindle cell component is separated by slitlike spaces containing extravasated erythrocytes and macrophages, with resulting hemosiderin deposits. An associated inflammatory component may be present, and this can become more prominent as lesions mature.
Intracellular eosinophilic globules, or red bodies, are present within the spindle cells and stain positively for phosphotungstic acid-hematoxylin, trichrome, and PAS stains. Although the cell of origin has not been clearly defined, vascular or lymphatic endothelium has been implicated. KS cells stain positively with factor VIII and ulex lectin; these results suggest an endothelial origin. The spindle cell component also stains positive with vimentin, CD31, and CD34, which are markers suggestive of a vascular endothelial origin. DNA from the human herpesvirus 8 (HHV8) has been identified in 100% of cases of AIDS-associated KS. HHV8 is thought to affect the expression of numerous proteins that influence cell regulation and survival, as well as the immune response; this suggests a role for the virus in the development of KS.
The classic form of KS is an indolent lesion associated with a very low mortality rate. Generally, patients with classic KS die from causes other than KS. Conversely, AIDS- or transplant-associated KS is a far more aggressive lesion with a high mortality rate that is associated with concurrent opportunistic infection and early visceral dissemination of KS lesions. KS lesions are divided into good and poor prognostic categories. Good prognostic features include lesions that are isolated to skin and lymph nodes with minimal mucosal disease, a CD4 count higher than 200, no evidence of thrush, and no systemic symptoms (eg, fever, night sweats, weight loss >10% of body weight).
Poor prognostic features include edematous or ulcerating lesions, a CD4 count lower than 200, the presence of thrush, a poor performance status, and the presence of other HIV-related illness. The mean survival time in patients with good prognostic factors is 30 months, while the mean survival in patients with poor prognostic factors is 7 months.
Although biopsy may be required to establish a diagnosis, the treatment for KS is nonsurgical and consists of either radiographic therapy (XRT) or chemotherapy. The choice of treatment is based on the extent of disease. Localized cutaneous disease, with or without regional nodal involvement, is managed with XRT. Patients with disseminated lymph node involvement, mucocutaneous involvement, or visceral involvement are treated with chemotherapy. Laser excision may be useful for palliation in cases of mucosal involvement or upper aerodigestive tract involvement. Recently, alfa-interferon and beta–human chorionic gonadotropin (HCG) have been shown to cause regression of lesions in a dose-dependent manner. In rapidly progressing KS, liposomal doxorubicin causes a 93% local response rate with fewer adverse effects, compared with other chemotherapy regimens, and liposomal doxorubicin is considered the treatment of choice in this situation.
Bone Sarcomas
Osteosarcoma
Osteosarcoma is the second most common skeletal neoplasm (multiple myeloma is the most common), with an incidence of 1 case per 100,000 people. Osteosarcomas account for as many as 5% of all head and neck primary tumors, and they are the most common bone sarcoma. Most osteosarcomas occur in the long bones of the limbs. Osteosarcoma involves the head and neck region in approximately 10% of reported cases. The mandible and maxilla are the most frequently affected sites, followed by the paranasal sinuses and skull. In long bones, incidence peaks in those aged 10-20 years, while head and neck cases occur in a slightly older population, with a peak incidence in those aged 20-40 years. Males are affected slightly more often than females.
Lesions frequently manifest as a swelling or mass over the jaw or cheek; pain and dental symptoms are less common. An association exists between development of osteosarcomas and a history of retinoblastoma (independent of any irradiation of this tumor), prior irradiation, Paget disease of bone, fibrous dysplasia, Li-Fraumeni syndrome, and chronic osteomyelitis. Osteosarcomas arising in the setting of Paget disease typically occur in those aged 60-70 years. The reported association with a history of retinoblastoma appears to be related to alterations in chromosome 13, the same chromosome implicated in retinoblastoma development. Serum alkaline phosphatase levels are elevated in 50% of patients with osteosarcoma. Elevated serum alkaline phosphatase levels in patients with any of the associated conditions may signal malignant transformation.
Radiographic studies demonstrate destructive lytic or sclerotic changes, which are sometimes associated with extension into adjacent soft tissue. Subperiosteal formation of new bone may occur adjacent to areas of bone loss. A sunburst pattern resulting from radiating spicules of bone has been described; however, this finding is not specific for osteosarcoma. Widening of the periodontal ligament may be present; this finding is not specific for osteosarcoma, but this finding is highly suggestive of malignancy.
At gross examination, tumors may appear soft and granular (osteolytic) or sclerotic and dense (osteosclerotic), depending on the degree of mineralization. Soft tissue extension is common. At histologic analysis, osteoid (a precursor of bone) is present within a sarcomatous stroma. The stromal cells may have anaplasia; their shape varies from spindled to round, and the cells contain hyperchromatic nuclei. The degree of vascularization varies considerably from scant to abundant.
The presence of osteoid is the distinguishing feature of this tumor, but osteoid may be absent in small, unrepresentative biopsy specimens. Osteoid is eosinophilic with hematoxylin-eosin staining and may resemble collagen when present in small quantities; immunohistochemical stains can help in differentiating the two. Unlike collagen, osteoid reacts positively with immunohistochemical stains for osteocalcin, a bone-specific protein produced by osteoblasts, and osteonectin, a bone-specific phosphorylated glycoprotein. Osteosarcomas also have strong alkaline phosphatase reactivity.
On the basis of the predominant component of the stroma, lesions can be subtyped as osteoblastic, chondroblastic, or fibroblastic. A giant cell–rich osteosarcoma subtype has been confirmed with osteocalcin staining. Osteoblastic tumors occur most frequently and have osteoclastic activity and increased vascularity. The prognosis is independent of the histologic subtype. Extraosseous osteosarcoma has been reported but is rare in the head and neck. Tumors are graded from stage I (well-differentiated, low grade) to stage IV (poorly differentiated, high grade) on the basis of increasing cellular atypia or anaplasia and increasing number of mitotic figures. In children, low-grade lesions are predominant.
Surgical excision is the main treatment for osteosarcoma. Cervical metastases are present in fewer than 10% of patients, and, when present, they usually result from direct extension rather than true lymphatic spread. Distant metastases are more common and occur in 33% of patients; most frequently, they involve the lungs. Local recurrence occurs in approximately 60% of patients, most commonly within the first year after treatment. Patients with extragnathic sites of involvement fare worse than those with gnathic sites. Reported mean 5-year survival rates are 43% for gnathic osteosarcoma and 9% for skull lesions, which are associated with a higher incidence of local recurrence and distant metastasis. Multifocal tumors are uniformly fatal. Patients with increased alkaline phosphatase levels appear to have a worse prognosis, as do patients with concomitant Paget disease.
Adjuvant radiation therapy has been used in the management of osteosarcoma when wide surgical margins cannot be obtained; however, osteosarcomas are relatively resistant to radiation therapy, and doses in excess of 6000 Gy are required. The use of neoadjuvant chemotherapy (cisplatin, doxorubicin) appears to be of benefit in some patients. A study by Mücke et al supported the efficacy of neoadjuvant chemotherapy. The study, which involved 36 patients with primary osteosarcoma of the craniomaxillofacial region, found that in patients treated with a combination of surgery and neoadjuvant chemotherapy, the 2- and 5-year overall survival rates were 100% and 66.7%, respectively, compared with 66.7% and 41.7%, respectively, in patients treated with surgery alone. [15]
Response to chemotherapy in osteosarcoma appears to be related to the expression of the p -glycoprotein multidrug resistance gene; patients with tumors that do not express this protein have a dramatic response to chemotherapy and markedly improved survival. Neoadjuvant chemotherapy is now recommended as part of a multimodality regimen for patients with osteosarcoma.
Alkaline phosphatase levels, when elevated preoperatively, can be used to monitor patients for recurrence after treatment.
Ewing sarcoma
Ewing sarcoma is a malignant primary bone tumor of primitive neuroectodermal derivation. Ewing sarcoma represents 4-7% of all primary bone tumors and is the second most common malignant bone tumor in children. Osseus (OES) and extraosseous (EOE) subtypes of Ewing sarcoma exist. OES accounts for most cases of Ewing sarcoma, and it has a predilection for the long bones (ie, femur, tibia, humerus) and pelvic girdle. Only 3% of cases of OES arise in the head and neck, where the calvaria and mandible are most frequently affected. [16, 17]
EOE accounts for 4-7% of all cases of Ewing sarcoma, and it most commonly affects the thigh, pelvis, and paravertebral soft tissues. The head and neck are the primary sites in 18% of cases of EOE. Most Ewing sarcomas occur in those aged 20 years or younger; more than 80% occur in patients aged 30 years or younger. Overall, a slight male predominance exists, but in head and neck sites, the sex distribution is equal. Metastasis is present in fewer than 20% of cases and primarily involves the lung; regional metastasis is uncommon.
The most common presenting feature in Ewing sarcoma is an enlarging mass or swelling; pain is present in approximately half of all patients. The etiology of these lesions is unknown, although a relationship between a history of prior irradiation or chemotherapy for childhood malignancies and subsequent development of Ewing sarcoma may exist. Radiologic studies characteristically demonstrate an osteolytic lesion and may show bone expansion, lamination, or cortical destruction; however, none of these findings is specific to Ewing sarcoma. Pathologic fractures may be present. CT scanning is considered superior to plain radiography in demonstrating disease extent and medullary involvement.
The pathologic appearances are identical for OES and EOE. On gross examination, Ewing sarcoma appears as a gray-white mass with areas of hemorrhage and necrosis. Histologically, Ewing sarcoma is a highly cellular lesion composed of small, densely packed cells with a low mitotic index and scant stroma. Cells are composed of uniformly appearing, small, round, hyperchromatic nuclei and scant cytoplasm; thus, it is designated a small, round, blue cell tumor. Prominent intracellular glycogen is present in Ewing sarcoma. On electron microscopy, neurosecretory granules can often be identified.
Results of immunohistochemical staining in Ewing sarcoma are positive with vimentin, HBA71, MIC2, and neuron-specific enolase; they are negative with muscle-specific actin, myogenin, and desmin. Despite the presence of neurosecretory granules, catecholamine secretion is not present. The cytogenetic translocation t(11;22)(q24;q12) is present in more than 90% of Ewing sarcomas. The MIC2 gene is expressed in more than 90% of Ewing sarcomas, as is the MIC2 gene glycoprotein product p30-32 and the CD99 antigen. These translocations appear in primitive neuroectodermal tumors (pNETs).
The t(11;22)(q24;q12) translocation, MIC2 gene, and its product p30-32 are also expressed in pNETs; this finding suggests that Ewing sarcoma is histogenetically related to pNET. Ewing sarcoma and pNET are likely related expressions of a spectrum of tumors, designated ES-pNET; Ewing sarcoma occurs most frequently and is the least differentiated tumor in this group.
Ewing sarcoma must be differentiated from other tumors that also have a small, round, blue cell appearance, in particular rhabdomyosarcoma and neuroblastoma. Rhabdomyosarcomas stain positive with muscle-specific actin, desmin, and myogen, and they do not express MIC2, unlike Ewing sarcoma. Neuroblastomas secrete catecholamines, stain positive with chromogranin, and do not have MIC2. The distinction between Ewing sarcoma and pNET is more difficult because of a lack of immunohistochemical markers that can be used to distinguish the two; an absence of neural differentiation supports a diagnosis of Ewing sarcoma.
Multimodality therapy for Ewing sarcoma is associated with markedly improved survival rates. Surgery followed by adjuvant XRT and multiagent chemotherapy dramatically improves survival rates, compared with single- or even dual-modality therapy. Complete surgical excision is undertaken whenever possible; nonoperative treatment is associated with a lower long-term survival rate than that of a treatment regimen that includes surgery. XRT has the risk of promoting the development of a second malignancy in young patients, and XRT may be withheld when complete surgical excision can be accomplished with clear margins. The use of adjuvant XRT is associated with improved local control because it treats microscopic residual disease. Multiagent chemotherapy has dramatically improved the 5-year survival rates from 10% prior to its use to 50-75% today. Ifosfamide with etoposide or vincristine, dactinomycin, and cyclophosphamide are most commonly used.
The prognosis appears to be dependent on the location of the primary tumor and the presence of distant metastasis at presentation. Survival rates in patients with Ewing sarcoma of the head and neck are significantly better than those of patients with tumors in other locations. Patients with cutaneous and subcutaneous variants of EOE are reported to have a very favorable prognosis. EOE is associated with a 5-year survival rate of approximately 70%. Survival rates for OES vary depending on the bone involved; gnathic sites are associated with the best survival rates, followed by long-bone involvement; patients with these have a better prognosis than those with pelvic involvement.
Involvement of the cervical vertebrae by OES is associated with an extremely poor prognosis. Overall 5-year survival rates for patients with OES are 54-74%. At presentation, metastasis is present in fewer than 20% of patients with Ewing sarcoma, which most commonly involves the lungs. The presence of pulmonary metastasis is associated with an average survival time of 10 months. Most treatment failures in patients who do not have distant metastases at presentation result from local recurrence.
Radiation-Induced Sarcomas
Ionizing radiation is known to damage chromosomes and chromosomal repair mechanisms. While irradiation is an important modality in the management of carcinoma, the radiation can induce a wide variety of cancers.
Determination of a causal relationship between prior irradiation and radiation-induced tumor formation requires that the following conditions are met: (1) radiation must have been delivered to the site in question, (2) the new malignancy must arise within the irradiated field, (3) the new tumor must be histologically distinct from the original primary lesion, and (4) a latent period between the time of radiation exposure and development of the new malignancy must be 5 years or longer.
Additionally, a causal relationship is supported when the difference in the incidence of the new malignancy in patients who have received irradiation and the incidence in a comparable control population is statistically significant.
Squamous cell carcinoma is the most common histologic type of radiation-induced malignancy. RISs account for 12% of postirradiation lesions, with an incidence of 0.03-0.3% in patients exposed to radiation, and they account for 0.5-5.5% of all sarcomas overall. Both osseous and soft tissue sarcomas can arise in previously irradiated tissues. Osseous sarcomas are more common and account for 63% of all RISs; osteosarcomas are most common, followed by malignant fibrous histiocytoma and fibrosarcoma. Malignant fibrous histiocytoma is the most common soft tissue RIS, followed by rhabdomyosarcoma, angiosarcoma, fibrosarcoma, chondrosarcoma, and leiomyosarcoma. Head and neck lesions account for 13-16% of RISs; no single site appears to have a predilection for the development of RIS.
A history of irradiation includes exposure to external-beam irradiation for various primary tumors and benign conditions and also exposure to radium 224 and thorium 232. Use of XRT in the management of malignancies in children is associated with a higher risk of RIS. RIS tends to occur at the edges of the radiation field, where the doses of radiation are sufficient to permanently alter a cell's ability to perform repair functions but insufficient to kill tumor cells. Adjuvant chemotherapy administered with XRT appears to increase the relative risk of RIS by a factor of 4 or more. The mean age of patients at occurrence is 48 years; no significant sex predilection has been reported.
The threshold dose for RIS is unknown; RIS has occurred after doses as low as 1340 cGy and doses as high as 16,440 cGy, with a mean delivered dose of 6000 cGy. The risk of RIS appears to increase with increasing radiation dose. Alterations in the p53 gene and the retinoblastoma Rb gene have been implicated in the development of RIS. The median latency period between irradiation and the diagnosis of RIS is 17 years, although a shorter latency period has been reported to occur among pediatric patients. An indirect relationship between the radiation dose and latency period has been described for radiation doses exceeding 4000 cGy, but this has not been consistently demonstrated.
RISs are associated with an outcome that is significantly poorer than that of stage-matched soft tissue and osteogenic sarcomas that arise independent of irradiation. Five-year disease-free survival rates for RIS are 10-30%; irradiation-independent sarcomas are associated with a 5-year survival rate of 54%. Prognosis is related to the location and resectability; thus, RIS of the extremities has a 30% 5-year survival rate, whereas RIS of the vertebral column and pelvis is associated with a 5-year survival rate lower than 5%. The survival rate of RIS of the head and neck is somewhere in between these rates.
Proposed explanations for the comparatively poor prognosis of RIS in the head and neck include the following factors: (1) delay in the diagnosis of disease in irradiated tissue, (2) compromised resection secondary to the proximity of the lesion to neurovascular structures, (3) limitation of treatment modalities in a previously irradiated field, (4) poor tumor sensitivity to chemotherapy, (5) locally aggressive behavior of RIS, and (6) host immunosuppression resulting from the combination of tumor related factors and previous treatment. In the setting of previous irradiation of the involved site, complete surgical excision is necessary to achieve meaningful survival. Adjuvant XRT and/or chemotherapy have a role in treatment, but the extent of adjuvant XRT is limited by the amount of radiation previously received.
-
Photomicrograph (hematoxylin-eosin) shows rhabdomyosarcoma with hypocellular and hypercellular areas. Round cell and spindle cell components are present. Spindle cells are marked by their fusiform shape and eosinophilic cytoplasm.
-
Photomicrograph shows rhabdomyosarcoma with characteristic positive actin staining.
-
Photomicrograph shows biphasic synovial sarcoma, which has fibrous and epithelioid components. Epithelioid cells line the cystic spaces, while fibrous cells span the intervening regions.
-
Photomicrograph depicts Ewing sarcoma as small round blue cell tumors. Empty spaces represent glycogen, which stains positively with periodic acid-Schiff (PAS) reagent.
-
Photomicrograph shows chondrosarcoma, which is marked by enlarged chondrocytes with nuclear pleomorphism and hyperchromasia. Binucleate or multinucleate cells may be present.
-
Photomicrograph shows malignant fibrous histiocytoma with a background of spindle cells in a cartwheel or storiform configuration, scattered histiocytes, and giant cells. An inflammatory infiltrate is often present.
-
Photomicrograph shows liposarcoma with lipoblasts of variable sizes and shapes. Lipoblasts can be either univacuolated or multivacuolated.
-
Photomicrograph shows fibrosarcoma with a fasciculated, or herringbone, pattern of spindle cells and an indistinct cell borders. The absence of giant cells is an important feature in distinguishing fibrosarcoma from malignant fibrous histiocytoma.
-
Photomicrograph shows hemangiopericytoma. Endothelium-lined vessels are compressed in a staghorn pattern and are surrounded by uniform small cells. Tumor cells are relatively uniform with little atypia and are external to the vascular reticulin layer.
-
Photomicrograph shows angiosarcoma. Pleomorphic and hyperchromatic endothelial cells with increased mitotic activity stack or pile up within the reticulin layer.









