Calcium Homeostasis
Calcium homeostasis is a complex process involving the following 4 key components: serum calcium, serum phosphate, 1,25-dihydroxyvitamin D-3, and parathyroid hormone (PTH). More than 99% of the total body calcium is stored in bone in the form of phosphate and hydroxide salts, predominantly as hydroxyapatite. Normally, a very small portion of this calcium is available for exchange in the serum.
A schematic diagram of calcium homeostasis can be seen below.
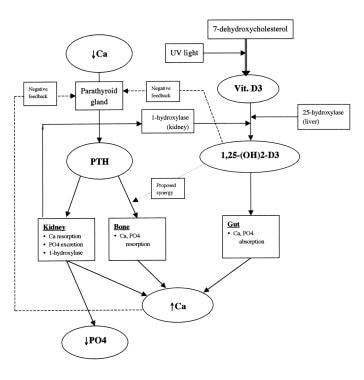 Schematic diagram of calcium homeostasis.
Schematic diagram of calcium homeostasis.
Parathyroid hormone (PTH)
Parathyroid hormone (PTH) is a polypeptide containing 84 amino acids that is secreted by the parathyroid glands after cleavage from preproparathyroid hormone (115 amino acids) to proparathyroid hormone (90 amino acids) to the mature hormone. The major target end organs for parathyroid hormone (PTH) action are the kidneys, skeletal system, and intestine.
The primary response to parathyroid hormone (PTH) by the kidney is to increase renal calcium resorption and phosphate excretion. In the kidney, parathyroid hormone (PTH) blocks reabsorption of phosphate in the proximal tubule while promoting calcium reabsorption in the ascending loop of Henle, distal tubule, and collecting tubule.
Parathyroid hormone (PTH) promotes absorption of calcium from the bone in 2 ways. The rapid phase brings about a rise in serum calcium within minutes and appears to occur at the level of the osteoblasts and osteocytes. Although it may seem counterintuitive that the cells that promote deposition of bone are involved in resorption, these cells form an interconnected network known as the osteocytic membrane overlying the bone matrix, but with a small layer of interposed fluid termed bone fluid. When parathyroid hormone (PTH) binds to receptors on these cells, the osteocytic membrane pumps calcium ions from the bone fluid into the extracellular fluid.
The slow phase of bone resorption occurs over several days and has 2 components. First, osteoclasts are activated to digest formed bone, and second, proliferation of osteoclasts occurs. Interestingly, mature osteoclasts lack parathyroid hormone (PTH) membrane receptors; activation and proliferation appear to be stimulated by cytokines released by activated osteoblasts and osteocytes or by differentiation of immature osteoclast precursors that possess parathyroid hormone (PTH) and vitamin D receptors.
The final important function of parathyroid hormone (PTH) is conversion of 25-hydroxyvitamin D to its most active metabolite, 1,25-dihydroxyvitamin D-3 [1,25-(OH)2 D3], by activation of the enzyme 1-hydroxylase in the proximal tubules of the kidney.
Feedback inhibition of parathyroid hormone (PTH) release occurs primarily by direct effect of calcium at the level of the parathyroid gland. Although not well elucidated, 1,25-(OH)2 D3 appears to exert a mild inhibitory effect on the parathyroid gland as well.
Vitamin D
Vitamin D-3 (cholecalciferol) is formed in the skin when a cholesterol precursor, 7-dehydroxycholesterol, is exposed to ultraviolet light. Activation occurs when the substance undergoes 25-hydroxylation in the liver and 1-hydroxylation in the kidney.
The primary action of 1,25-(OH)2 D3 is to promote gut absorption of calcium by stimulating formation of calcium-binding protein within the intestinal epithelial cells. Vitamin D also promotes intestinal absorption of phosphate ion, although the exact mechanism is unclear. Negatively charged phosphate ion may passively flow through the intestinal cell because of flux of the positively charged calcium ion. In bone, vitamin D may play a synergistic role with parathyroid hormone (PTH) in stimulating osteoclast proliferation and bone resorption.
Compared to parathyroid hormone (PTH), vitamin D exerts a much slower regulatory effect on calcium balance.
Disorders of Parathyroid Hormone Metabolism
Hyperparathyroidism
Primary hyperparathyroidism (HPT) is defined as abnormal hypersecretion of parathyroid hormone (PTH), producing hypercalcemia and hypophosphatemia. In the face of persistent hypercalcemia, parathyroid hormone levels should be low due to feedback inhibition of the parathyroid glands. Thus, even high normal parathyroid hormone levels are considered pathologic in patients with chronic hypercalcemia.
Primary hyperparathyroidism (HPT) is the most common cause of elevated parathyroid hormone (PTH) and calcium levels. Approximately 85% of cases are found to be caused by an isolated parathyroid adenoma, 15% by diffuse parathyroid hyperplasia, and less than 1% by parathyroid carcinoma. Other causes include neck or mediastinal parathyroid cysts, which are also uncommon. Rarely, primary hyperparathyroidism (HPT) may be related to multiple endocrine neoplasia (MEN); family history or other endocrine tumor warrants screening for MEN.
MEN syndromes have an autosomal dominant hereditary pattern with a predilection to develop tumors of the endocrine organs, including the parathyroid glands. Parathyroid hyperplasia is seen in both MEN 1 and MEN 2a. MEN 1 is characterized by parathyroid hyperplasia, pancreatic islet cell tumors, and pituitary adenomas. MEN 2a includes parathyroid hyperplasia, medullary carcinoma of the thyroid, and pheochromocytoma.
A prospective cohort study by Vaidya indicated that a connection exists between low physical activity and the development of primary hyperparathyroidism. The study, which included 69,621 females, found that the age-adjusted relative risk of developing primary hyperparathyroidism was reduced by 50% in women in the highest physical activity quintile, compared with women in the lowest quintile. The investigators also found that, compared with participants with high physical activity and high calcium intake, the adjusted relative risk of developing the condition was 2.37-fold greater in women with a combination of low physical activity and low calcium intake. [1]
Incidence of primary hyperparathyroidism (HPT) in the United States has been reported as 5-50 per 10,000 individuals. The disease is common in individuals older than 40 years and has a female-to-male ratio of 3:1.
Secondary hyperparathyroidism (HPT) is a compensatory hyperfunctioning of the parathyroid glands caused by hypocalcemia or peripheral resistance to parathyroid hormone. As opposed to primary hyperparathyroidism (HPT), treating the underlying cause can reverse secondary hyperparathyroidism (HPT). The most common setting is in a patient with end-organ failure from chronic renal insufficiency, with hypocalcemia and hyperphosphatemia. Advanced age and malnutrition are risk factors for developing secondary hyperparathyroidism in patients with chronic kidney disease. Less commonly, secondary hyperparathyroidism may be caused by calcium malabsorption, osteomalacia, vitamin D deficiency, or deranged vitamin D metabolism.
Tertiary hyperparathyroidism (HPT) occurs in a setting of previous secondary hyperparathyroidism (HPT) in which the glandular hyperfunction and hypersecretion continue despite correction of the underlying abnormality, as in renal transplantation.
Hyperparathyroidism in pregnancy is a rare condition that can place the fetus at significant risk. Maternal calcium crosses the placenta resulting in chronic suppression of fetal parathyroid glands. Following delivery, neonatal serum calcium drops without maternal calcium provided through the placenta and profound perinatal hypocalcemia ensues. Neonatal parathyroid function returns in approximately 7-10 days. Current treatment recommendations include exploration with total parathyroidectomy and autotransplantation during the second trimester if possible.
Familial hypocalciuric hypercalcemia (FHH) is a disease with an autosomal dominant mode of inheritance linked to a defect on chromosome 3. The exact mechanism of the disease is not known, but it appears that affected individuals have an abnormal calcium sensor. Parathyroid hormone levels may be elevated because of abnormal calcium detection. Because renal calcium resorption in these patients is greater than 99%, diagnosis is made by a calcium-to-creatinine clearance ratio of less than 0.010, while in those with primary hyperparathyroidism (HPT) the ratio is 0.015-0.040. Patients should have a family history positive for hypercalcemia. Surgery is not indicated in these individuals.
Hypoparathyroidism
Hypoparathyroidism is an uncommon congenital or acquired condition in which parathyroid hormone secretion is deficient or absent. [2, 3, 4] Hypocalcemia and hyperphosphatemia are usually present. By far, hypoparathyroidism most commonly results from an iatrogenic cause; it usually follows parathyroid surgery or total thyroidectomy. An Italian study, by Marcucci et al, found that 67.6% of cases of chronic hypoparathyroidism occurred postsurgically, with the next highest percentage of such cases (14.6%) being idiopathic. [5]
A retrospective study by Nawrot et al found the prevalence of permanent hypoparathyroidism (ie, hypoparathyroidism lasting more than 12 months) following total thyroidectomy to be 20.2%, versus 6.7% following near-total thyroidectomy and 4.2% after subtotal thyroidectomy. The overall rate of permanent hypoparathyroidism among the study’s 401 patients was 8.5%. [6]
Abnormalities of the third and fourth branchial pouches, such as DiGeorge syndrome, may lead to agenesis or dysgenesis of the parathyroid glands as well as athymia. Isolated familial cases of metabolic hypoparathyroidism have been reported. Some cases of hypoparathyroidism categorized as idiopathic may have an autoimmune basis and other endocrine deficiencies; T-cell dysfunction also may be involved.
A study by Almquist et al indicated that in patients who undergo total thyroidectomy, those who consequently develop permanent hypoparathyroidism have a greater mortality risk than do the other patients. According to the report, which had a mean follow-up period of 4.4 years, the adjusted hazard ratio for death in total thyroidectomy patients with permanent hypoparathyroidism was 2.09, compared with the rest of the study’s cohort. [7]
A study by Lončar et al indicated that following total or completion thyroidectomy, a reduction in parathyroid hormone levels of greater than 70% reliably predicts the occurrence of persistent hypoparathyroidism. [8]
Guidelines
In 2022, the Second International Workshop on Parathyroid Disorders published guidelines on the evaluation and management of hypoparathyroidism. These include, but are not limited to, the following [9] :
-
Postsurgical hypoparathyroidism that persists for more than 12 months may be considered permanent
-
In order to predict whether permanent postsurgical hypoparathyroidism will not develop, it is recommended that serum parathyroid hormone (PTH) be evaluated within 12-24 hours after total thyroidectomy; long-term hypoparathyroidism is virtually excluded if the PTH level is above 10 pg/mL (1.05 pmol/L)
-
Genetic testing may be useful in persons with nonsurgical hypoparathyroidism if the patient has a positive family history of nonsurgical hypoparathyroidism, if syndromic features are present, or if the individual is younger than age 40 years
-
Complications associated with hypoparathyroidism include nephrocalcinosis, nephrolithiasis, renal insufficiency, cataracts, seizures, cardiac arrhythmias, ischemic heart disease, depression, and an increased infection risk; patients must be carefully evaluated and laboratory indices must be closely monitored in order to minimize the complications
Differential diagnoses of hypercalcemia
See the list below:
-
Parathyroid
Primary hyperparathyroidism (HPT)
Solitary adenoma
Parathyroid hyperplasia
Multiple endocrine neoplasia
Parathyroid carcinoma
Neck or mediastinal parathyroid cyst
Lithium therapy
-
Malignancy
Solid tumor with metastases (ie, breast)
Solid tumor with parathyroid-related peptide production (ie, lung, kidney)
Hematologic malignancy (multiple myeloma, lymphoma, leukemia)
-
Vitamin D
Vitamin D intoxication
Sarcoidosis or other granulomatous diseases, which cause increased 1,25-(OH)2 D3
-
Idiopathic hypercalcemia of infancy
-
High bone turnover
Hyperthyroidism
Immobilization
Vitamin A intoxication
-
Renal
Secondary hyperparathyroidism (HPT)
Familial hypocalciuric hypercalcemia
Thiazide diuretics
Aluminum intoxication
Milk-alkali syndrome
Pathophysiology
The exact cause of parathyroid adenoma is unknown. Single adenoma is the most common cause of hyperparathyroidism and may affect superior and inferior parathyroid glands. Histologically, adenomas are hypercellular and have very little fat when compared with normal glandular tissue. Fat content tends to increase with age (until age 25-30 years), thus comparison to a normal gland is important to ensure the diagnosis is adenoma. The adenoma may also be surrounded by a rim of normal parathyroid tissue. Chief cells, which have regular central nuclei, predominate. Foci of larger acidophilic oxyphilic cells and clear cells are present. Adenomas may have clear cell variants.
Primary parathyroid hyperplasia is defined as proliferation of the parenchymal cells leading to an increase in gland weight in multiple parathyroid glands in the absence of a known stimulus for parathyroid hormone secretion. Two types of parathyroid hyperplasia exist: chief cell hyperplasia and water cell or clear cell hyperplasia.
In primary hyperplasia, the most common finding is chief cell hyperplasia, although clear cell variants can rarely occur. Again, stromal fat is decreased in proportion to the magnitude of hyperplasia. A differentiating feature seen in approximately one third of adenomas but not in hyperplasia is a well-defined capsule separating the adenoma from normal parathyroid tissue.
Distinguishing normal-functioning parathyroid glands from hyperfunctioning parathyroid glands visually is difficult.
Clinical Presentation
Hyperparathyroidism
The mnemonic "painful bones, psychic moans, abdominal groans, and renal stones" has become largely historical because most patients present long before the disease progresses to multiple-system involvement. In the United States, the most common presentation of hyperparathyroidism (HPT) is an elevated serum calcium level reported on routine screening by a primary care physician and has been termed asymptomatic hyperparathyroidism. In the absence of overt abnormalities, one must be cautious as symptoms may be subtle.
Another common scenario (15-20%) occurs when a patient presents with a calcium oxalate kidney stone and is found to be hypercalcemic (4% incidence with this type of stone). As the mnemonic suggests, clinical manifestations of hypercalcemia may be described by the following organ systems:
-
Kidney/urinary tract: Kidney stones, polyuria, nocturia, renal colic
-
Skeletal system: Bone loss, osteitis fibrosis cystica [10]
-
Neuromuscular: Muscle weakness, fatigue/malaise
-
Neurologic: Depression, nervousness, cognitive dysfunction, psychosis, confusion, headache
-
Gastrointestinal: Peptic disease, pancreatitis, cholelithiasis, nausea, vomiting, loss of appetite, constipation, abdominal pain
-
Cardiovascular: Hypertension, arrhythmias
However, on careful questioning of an asymptomatic patient, one often can elicit subtle symptoms, such as nonspecific fatigue, weakness, musculoskeletal complaints, constipation, depression, or possibly a history of peptic ulcer, hypertension, cholelithiasis, pancreatitis, and gout or pseudogout. Additionally, prolonged hypercalcemia can lead to metastatic calcifications in blood vessels, soft tissues, and joints (chondrocalcinosis).
The most common conduction abnormality observed with hypercalcemia is a shortened QT interval, which should be documented with an electrocardiogram.
Calciphylaxis is a rare, but disastrous, dermatologic complication of hypercalcemia that occurs most frequently in patients with secondary hyperparathyroidism (HPT) and end-stage renal disease. Skin lesions begin as pink or mottled purpura that progress to ulceration and necrosis. Secondary infection and sepsis can develop and lead to mortality rates of greater than 80%. The exact mechanism is not known but is likely related to microvascular occlusion.
Osteitis fibrosa cystica, in which subperiosteal bone resorption is followed by the formation of cysts that may distort bony architecture, is a rare presentation. The most commonly involved bones are the phalanges and distal clavicles; involvement of the mandible is less likely, and the maxilla is rarely affected. If hemorrhage into a cyst occurs, it is followed by giant cell reparative granulomas known as brown tumors. Diagnosis is confirmed by an elevated serum level of intact parathyroid hormone or high normal parathyroid hormone in the setting of hypercalcemia.
Hypoparathyroidism
Patients with hypoparathyroidism present with hypocalcemia, mental changes, and neuromuscular excitability or tetany. Anatomic abnormalities, though not readily apparent, include intracranial calcifications and cataract formation. Young children may have disturbed dentition.
Pseudohypoparathyroidism is a rare hereditary disorder caused by end-organ failure that negatively impacts the ability to appropriately detect and respond to parathyroid hormone (PTH). Most patients with this disorder have a characteristic phenotype of short stature, round face, brachydactyly, and heterotopic calcification. Low serum calcium and high phosphate levels are accompanied by a high parathyroid hormone (PTH) level. Genetically, all patients have abnormalities at the GNAS cluster on chromosome 20q13.3, and research for specific alterations is ongoing. [11]
Medical Therapy of Primary Hyperparathyroidism
Severe hypercalcemia should be managed promptly with administration of intravenous fluids and a loop diuretic to block calcium resorption. Calcitonin (2-8 U/kg IV/IM/SC q6-12h) can lower serum calcium levels by inhibiting bone resorption, but tachyphylaxis occurs quickly. Bisphosphonates also inhibit bone resorption, but electrolyte abnormalities such as hyperphosphatemia or hypophosphatemia, hypocalcemia, and hypomagnesemia are common. Calcimimetics, such as cinacalcet (Sensipar), mimic circulating calcium in the blood in an attempt to cause the parathyroid glands to release less parathyroid hormone. This may be an option for patients with hyperparathyroidism who have failed surgical therapy or are not surgical candidates.
Primary hyperparathyroidism (HPT) is usually managed surgically. Observation of primary hyperparathyroidism is reserved mainly for patients with comorbid conditions who cannot tolerate surgery or for very elderly patients who may die of another cause. In postmenopausal women with hyperparathyroidism, estrogen supplements and alendronate may help maintain bone density. Patients who do not undergo parathyroidectomy should be monitored at regular intervals with metabolic panels and assessment of bone mass (dual-energy radiographic absorptiometry [DRA] scan, bone mineral density).
Surgical Treatment of Hyperparathyroidism
Parathyroidectomy is indicated for most patients with primary hyperparathyroidism (HPT) and for those with confirmed tertiary hyperparathyroidism, particularly in healthy patients younger than 50 years. Patients with secondary hyperparathyroidism may require parathyroidectomy if medical therapy fails to control calcium levels or if complications, such as calciphylaxis, develop. In primary hyperparathyroidism, surgery is always recommended for symptomatic patients and has been linked to improved bone health, reduced fracture rates, and improved cognition. Surgical indications in asymptomatic patients were put forth from a consensus conference held by the National Institutes of Health in 1990 and have since undergone three revisions, most recently in 2013; [12] they include the following:
-
Elevation of serum calcium more than 1 mg/dL above the upper limit of the reference range for the laboratory
-
Patient request for surgery or those who may be unsuitable for long-term monitoring
-
Glomerular filtration rate (GFR) below 60 mL/min and/or 24-hour urine calcium above 400 mg/d and increased kidney stone risk by biochemical analysis
-
Presence of kidney stones by history or radiography
-
Reduction of bone mass more than 2.5 standard deviations below normal by one of several noninvasive methods of measuring bone mass and/or radiographic evidence of vertebral fracture
-
Age younger than 50 years
Removal of the abnormal gland is indicated for adenoma, and resection of 3.5 glands is the standard treatment for 4-gland hyperplasia. For a detailed discussion of surgical anatomy and technique, please refer to the article Minimally Invasive Surgery of the Parathyroid.
Preoperative evaluation
For a discussion of preoperative localization studies for parathyroid adenoma and their impact on operative management, please refer to the article Minimally Invasive Surgery of the Parathyroid.
Preoperative screening should include a serum calcium level and a confirmed elevated or inappropriately high normal intact parathyroid hormone level. Intact parathyroid hormone assay differentiates primary hyperparathyroidism (HPT) from hypercalcemia of malignancy because the parathyroid-related peptide secreted from some tumors is a much larger protein, which shares a 50% homology with the 36 N -terminal amino acids of parathyroid hormone. Many endocrinologists also recommend a 24-hour calcium collection for calculation of the calcium-to-creatinine clearance ratio to exclude familial hypocalciuric hypercalcemia (FHH).
Intraoperative parathyroid hormone monitoring
Parathyroid hormone (PTH) has a circulating half-life of between 2-5 minutes, making intraoperative measurement extremely useful for confirming removal of hyperfunctioning tissue. A baseline serum parathyroid hormone is drawn before excision and a second level is taken 10-20 minutes after gland removal; several criteria have been proposed, but in general a decrease of 50% indicates successful extirpation of all hyperfunctioning tissue. [13, 14] Caution is prudent, however, as specificity values for this cutoff level range from 70-90%, and laboratory data should be interpreted on a case-by-case basis. Convincing laboratory evidence of successful removal obviates the need for four-gland exploration and saves significant operating time, cost, and potential morbidity. [15, 16]
Postoperative care
Calcium levels should be followed postoperatively. Because of chronic feedback suppression of normal parathyroid tissue, short-term hypocalcemia can be expected following most parathyroidectomies and is generally left untreated unless serum calcium is markedly decreased or the patient becomes symptomatic; some favor empiric treatment with oral vitamin D-3 and calcium.
Hungry bone syndrome is a condition that may follow parathyroidectomy; it is marked by hypocalcemia and hypophosphatemia. It is usually seen in patients with long-standing hyperparathyroidism (HPT) and extensive bone resorption. The proposed mechanism is a rebound uptake of calcium and phosphorus by bones, which have long been starved of these metabolites. If symptomatic, treatment is IV calcium.
Follow-up
Serum calcium should be monitored several weeks after surgery until calcium levels have stabilized. Calcium and parathyroid hormone (PTH) are checked 6 months postoperatively to exclude persistent hyperparathyroidism (HPT). Disruption of vascular supply may lead to hypoparathyroidism, particularly when all four glands are dissected or a gland is autotransplanted. Patients with hypoparathyroidism are treated with oral calcium and vitamin D; intravenous calcium is reserved for severe or symptomatic hypocalcemia. When at least one parathyroid gland is identified and preserved, parathyroid hormone levels usually return to normal, but four-gland removal can lead to permanent hypoparathyroidism.
Complications and Outcomes of Surgery
Complications
Temporary hypocalcemia is a common consequence of parathyroidectomy. Other short-term complications of parathyroidectomy include true vocal fold paralysis/paresis (due to subtle, recurrent laryngeal nerve injury), bleeding, hematoma, and, rarely, wound infection.
Persistent hypoparathyroidism is an uncommon consequence that requires long-term calcium and vitamin D-3 supplementation. Severe, recurrent laryngeal nerve injury resulting in permanent paralysis may be well compensated over time, but voice complaints or aspiration may warrant vocal fold medialization.
Outcome and prognosis
Cure rate following initial parathyroidectomy is 95-97%. Reexploration for recurrent or persistent hypercalcemia has a slightly lower success rate of 85-90%.
Future and Controversies
Radio-guided parathyroidectomy is being performed at an increasing number of centers in the United States. The radiotracer is injected 1.5-3 hours before surgery to allow for thyroid washout. Through a small incision, the surgeon uses a gamma probe to locate the abnormal gland, keeping dissection and blood loss to a minimum. This promising technique offers other potential advantages to the patient, including the option of local anesthesia with sedation and outpatient management. The gamma probe can be an invaluable tool to reduce the time required to locate an adenoma in patients who have undergone previous excision. The radio-guided technique has also proven useful for surgical treatment of secondary and tertiary hyperparathyroidism. [17, 18]
Therapeutic angiographic embolization also has been reported with immediate decrease in venous parathyroid hormone (PTH). One series of 24 patients with persistent hyperparathyroidism (HPT) following surgery reported a long-term success rate of 71%. This technique may also be useful for ectopic adenomas that are located in anatomic regions that are less accessible to or at higher risk with surgical approaches. [19]
Recent identification and cloning of the calcium sensor in the parathyroid gland has opened speculation into pharmacologic inhibition of parathyroid hormone (PTH) release. A calcium sensor–activing drug has been studied in a phase 2 trial for secondary hyperparathyroidism. [20] Until such a drug is approved, however, primary hyperparathyroidism (HPT) remains a surgical disease.
Anatomy
For a detailed discussion of the anatomy of parathyroid glands, please refer to the article Minimally Invasive Surgery of the Parathyroid.
-
Schematic diagram of calcium homeostasis.





