Practice Essentials
Congenital anomalies of the esophagus comprise a diverse group of malformations. This chapter discusses embryology of the developing esophagus and esophageal anomalies secondary to its aberrant development.
The chapter also reviews major esophageal malformations as follows:
-
Esophageal atresia and tracheoesophageal fistula
-
Laryngotracheoesophageal cleft
-
Esophageal stenosis and webs
-
Foregut duplications
-
Congenital bronchopulmonary foregut malformations
-
Diverticulum of esophagus
-
Congenital short esophagus
An image depicting common types of esophageal atresia and/or tracheoesophageal fistula can be seen below.
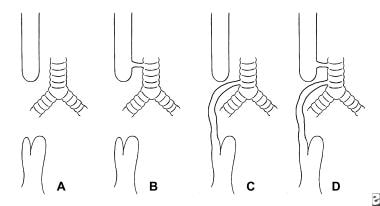 Common types of esophageal atresia include (A) pure atresia of the esophagus, (B) esophageal atresia with proximal tracheoesophageal (TE) fistula, (C) esophageal atresia with distal TE fistula, and (D) esophageal atresia with double fistula.
Common types of esophageal atresia include (A) pure atresia of the esophagus, (B) esophageal atresia with proximal tracheoesophageal (TE) fistula, (C) esophageal atresia with distal TE fistula, and (D) esophageal atresia with double fistula.
Signs and symptoms of congenital malformation of the esophagus
These include the following:
-
Esophageal atresia and tracheoesophageal (TE) fistula - In the neonate, excessive drooling is often the first symptom; the baby often chokes, regurgitates, and becomes cyanotic, during feedings; overflow of pharyngeal secretions into the trachea results in noisy breathing
-
Esophageal atresia without fistula - In the neonatal period, infants usually present with excessive drooling and a scaphoid abdomen
-
TE fistula without atresia - Presentation symptoms can range from coughing and choking spells (precipitated by feeding), to abdominal distention due to passage of air from the fistula, to recurrent and severe pneumonias that often are unresponsive to antibiotics
-
Laryngotracheoesophageal cleft - Clinicians may be alerted by feeding difficulties, husky cry, or aspiration pneumonia; stridor, coughing, and cyanotic episodes precipitated by feeding are all symptoms that may vary in severity depending on cleft extent
-
Esophageal stenosis and webs - Patients may present with aspiration and recurrent pneumonia; once solid food is introduced, dysphagia and regurgitation become obvious
-
Foregut duplications - Large duplications can result in cardiac or respiratory symptoms; neurologic symptoms often predominate if there are associated vertebral anomalies
-
Congenital bronchopulmonary foregut malformations - Symptoms include respiratory distress, cough during feeding, and recurrent pneumonias
Diagnosis of congenital malformations of the esophagus
This includes the following:
-
Esophageal atresia and TE fistula - Diagnosis of esophageal atresia is confirmed by inability to pass a firm oral or nasal tube into the stomach
-
Esophageal atresia without fistula - Radiologic findings pathognomonic of pure esophageal atresia include a dilated upper pouch and absence of air below the diaphragm
-
TE fistula without atresia - Bronchoscopy remains the investigation of choice to confirm diagnosis
-
Laryngotracheoesophageal cleft - Definite diagnosis is made endoscopically; suspension microlaryngoscopy is necessary to avoid missing the subtle defect
-
Esophageal stenosis and webs - Endoscopy with biopsy and pH monitoring of the esophagus help to eliminate the possibility of a stricture secondary to gastroesophageal reflux; barium swallow demonstrates narrowing of the distal esophagus
-
Foregut duplications - Chest radiographs, ultrasonograms, barium contrast studies, computed tomography (CT) scans, and nuclear (technetium) scans can contribute to the evaluation
-
Congenital bronchopulmonary foregut malformations - Chest radiographs, ultrasonography, barium contrast studies, CT scans, and magnetic resonance imaging (MRI) can contribute to evaluation
Management of congenital malformations of the esophagus
This includes the following:
-
Esophageal atresia and TE fistula - Primary repair is the treatment of choice for esophageal atresia and TE fistula; the fistula is first divided and closed near the trachea to avoid formation of a tracheal diverticulum; the proximal end of the esophagus then is mobilized until the anastomosis can be performed with acceptable tension
-
Esophageal atresia without fistula - Continuous suction of the upper pouch and a feeding gastrostomy constitute initial treatment; the patient then is followed closely with contrast radiographs until primary esophagus repair can be performed, usually at an age of 2-3 months.
-
Tracheoesophageal fistula without atresia - Once identified, the fistula is divided and oversewn; endoscopic obliteration of the fistula has been attempted with various techniques, such as tissue adhesives, electrocautery, and sclerosants
-
Laryngotracheoesophageal cleft - Type I (Benjamin and Inglis classification) is usually corrected with growth; types II and III can be corrected through cervical incision, while type IV requires lateral thoracotomy incision combined with a cervical approach; others require an anterior transtracheal repair
-
Esophageal stenosis and webs - Pneumatic dilatation under fluoroscopy may be diagnostic and therapeutic; while it expands and cures a fibromuscular stenosis, a persistent "waist" in the balloon indicates a cartilaginous ring and the necessity for resection; laser lysis of webs has been described and may be attempted in selected cases
-
Foregut duplications - Definitive treatment involves surgical resection of the duplication
-
Congenital bronchopulmonary foregut malformations - Definitive treatment is surgical
Embryology of the Esophagus
Development of the esophagus occurs during embryonic and fetal periods.
Embryonic period
The embryonic period extends from conception to the ninth week of gestation. During the latter half of the third week of development, the primitive foregut develops a ventral diverticulum that is cranial to the hepatic primordium and caudal to the fourth and fifth pharyngeal pouches. The diverticulum grows caudally and develops bronchopulmonary buds soon after appearance. The trachea develops from further caudal growth of the respiratory diverticulum.
During the fourth and fifth weeks of development, the rapidly growing heart and liver stretch the esophagus. Because of the stretching, the esophagus narrows almost to obliteration at the level of the carina. Although vacuoles have been seen within the esophagus during this period, the lumen of the esophagus remains intact. In addition, the bronchial primordia curve in a dorsal direction, likely due to the growth of the pericardium anteriorly. The dorsal "embracement" of the esophagus results in close approximation of the tracheal bifurcation to the front wall of the esophagus, further narrowing its lumen.
Between the sixth and eighth weeks of gestation, the epithelium becomes 2-5 cells thick and remains stratified columnar epithelium. The esophagus is also surrounded by a layer of undifferentiated mesenchyme and a circular layer of myoblasts. Longitudinal muscle fibers appear in the lower esophagus as the circular layer of muscle becomes well established. Cartilage also appears in the tracheobronchial tree.
Fetal period
The fetal period encompasses the ninth week of gestation until birth. During the 10th week of development, stratified columnar epithelium becomes ciliated, and mesenchymal ridges result in longitudinal folds of mucosa. Muscular proliferation peaks during the 11th and 12th weeks, and epithelial and mesenchymal proliferation decrease from the 10th to 15th week. Ganglion cells also appear in the myenteric plexus, while the longitudinal muscle becomes well defined between the 10th and 12th week of gestation. In addition, the epithelium is completely ciliated, and proliferation occurs only in the basal layers at the 12th week.
Striated muscle appears in the upper esophagus from the 12th to 15th week. Furthermore, the muscularis mucosa becomes well defined, and typical mucosal folds formed by longitudinal mesenchymal ridges can be appreciated.
During the fourth and fifth months of gestation, stratified squamous epithelium replaces the ciliated columnar epithelium. Growth of the esophagus continues at a slower pace once morphological changes conclude. On a functional level, swallowing first appears at the 14th week and is well established by the end of the fourth month of gestation.
Esophageal Atresia and Tracheoesophageal Fistula
Esophageal atresia is improper development of the esophagus, typically in the sense that the upper esophagus does not connect to the lower esophagus and stomach. A tracheoesophageal (TE) fistula occurs when the esophagus connects to the trachea.
Incidence
The literature variably places incidence of esophageal atresia at 1 per 3000-4500 live births. Etiology has been attributed to genetic factors, infections, and teratogens, but in most instances, no cause is identifiable. Oddsberg et al (2008) published a Swedish nationwide, population-based, case-control study of 2,305,858 deliveries, 722 cases of EA and 3610 controls. [1] They concluded that children of mothers who are having their first delivery, are of older age, and are of white ethnicity are at an increased risk of esophageal atresia.
Classification
Esophageal atresia and/or TE fistula have been classified anatomically into 5 different types, as follows:
-
Pure atresia of the esophagus (7.7%)
-
Esophageal atresia with proximal TE fistula (0.8%)
-
Esophageal atresia with distal TE fistula (86.5%)
-
Esophageal atresia with proximal and distal fistula (0.7%)
-
H-type fistula (4.4%)
Associated anomalies
Approximately half of babies with esophageal atresia have other associated congenital malformations (see the image below). Cardiac malformations occur in up to 28% of patients. The most common cardiac anomalies are patent ductus arteriosus, ventricular septal defect, atrial septal defect, and right aortic arch. Other gastrointestinal malformations also can occur, with an imperforate anus most frequent (10%).
Common types of esophageal atresia include (A) pure atresia of the esophagus, (B) esophageal atresia with proximal tracheoesophageal (TE) fistula, (C) esophageal atresia with distal TE fistula, and (D) esophageal atresia with double fistula.
Anomalies of the musculoskeletal system include hemivertebrae, rib malformations, and limb anomalies. Association with anomalies or malformations of the vertebral, anal, cardiac, tracheal, esophageal, renal, and limb organs (VACTERL) has been observed in 10% of patients with esophageal atresia.
Other anomalies associated with esophageal atresia include pulmonary hypoplasia, pulmonary sequestration, congenital diaphragmatic hernia, tracheal atresia, and chromosomal anomalies.
Diagnosis and clinical findings
During gestation, a diagnosis of esophageal atresia usually can be made using fetal ultrasound. Almost all patients with esophageal atresia (and up to 60% of patients with atresia and TE fistula) have polyhydramnios.
In the neonate, excessive drooling is often the first symptom. If feedings are attempted, the baby often chokes, regurgitates, and becomes cyanotic. Overflow of pharyngeal secretions into the trachea results in noisy breathing, and respiratory distress is progressive. If mechanical ventilation is necessary and lung compliance is poor, massive abdominal distention resulting in gastric rupture may occur due to free passage of air through the fistula into the stomach.
Diagnosis of esophageal atresia is confirmed by inability to pass a firm oral or nasal tube into the stomach. Perform a radiograph to confirm the position of the tube. Also, perform a complete physical examination to assess cardiopulmonary status and to seek other congenital anomalies.
Management
The goal of treatment is to surgically restore esophageal continuity as soon as the baby can tolerate the procedure. If respiratory status is inadequate, or if further investigation is required to assess for other congenital anomalies, surgery may be delayed for a few days postpartum to avoid aspiration pneumonia.
In premature infants with respiratory distress syndrome, a gastrostomy and fistula division can be performed. This decompresses the abdomen and allows adequate lung ventilation. Once the infant is stabilized and more mature, a definitive procedure to restore esophageal continuity can be undertaken.
Primary repair is the treatment of choice for esophageal atresia and TE fistula. The fistula is first divided and closed near the trachea to avoid formation of a tracheal diverticulum. The proximal end of the esophagus then is mobilized until the anastomosis can be performed with acceptable tension. This is performed with single-layer, interrupted, synthetic resorbable suture.
A multi-institutional study by Okuyama et al indicated that thoracoscopic repair of esophageal atresia/TE fistula produces outcomes comparable to those of open surgical repair. The investigators reported that the thoracoscopic procedure was successful in 52 of 58 patients (89.7%) who underwent the operation, with the remaining six patients requiring conversion to open thoracotomy because of the presence of a long gap or right aortic arch or because of intraoperative instability. Three of the 58 patients (5.2%) developed recurrent TE fistula, and 13 patients (22.4%) needed subsequent fundoplication. [2]
In a study of children with Gross type C esophageal atresia (esophageal atresia with a distal TE fistula), Kambe et al indicated that while early delivery, low body weight, and a long esophageal gap are reportedly fundoplication risk factors, others include polyhydramnios and prenatal diagnosis of esophageal atresia. The investigators said that these last factors may induce premature delivery and that, therefore, when esophageal atresia is suspected, polyhydramnios should be managed in order to avoid early delivery. [3]
A literature review by Connor et al examined common long-term problems in patients over age 10 years who had previously undergone esophageal atresia repair; rates, based on pooled estimated prevalence, were as follows [4] :
-
Dysphagia (50.3%)
-
Gastroesophageal reflux disease without histologic esophagitis (56.5%)
-
Gastroesophageal reflux disease with histologic esophagitis (40.2%)
-
Wheeze (34.7%)
-
Recurrent respiratory tract infections (24.1%)
-
Physician-diagnosed asthma (22.3%)
-
Persistent cough (14.6%)
-
Barrett esophagus (6.4%): Compared with the general population, four times the rate of occurrence in adults and 26 times the rate in children
A German study, by Hölscher et al, indicated that in many cases, a greater amount of aftercare and support than previously thought are required by patients and families in the first year after primary reconstruction for esophageal atresia. Among the findings, 59% of patients reportedly suffered gastroesophageal reflux symptoms, with fundoplication required in 33% of cases. Moreover, 68% of patients needed regular bougienage of anastomotic strictures, with repeat dilatations required by 36% in the first postoperative year. Sixty-six percent of patients needed postsurgical enteral nutrition by way of a nasogastric tube, with the tube required until the sixth week of life in 40% of patients. Nutritional support was needed in 25% of patients more than 1 year postsurgery. In addition, more than 50% of parents believed that they had not been sufficiently advised regarding long-term morbidities in their children. [5]
A study by Dittrich et al indicated that even after repair, esophageal atresia significantly affects long-term pulmonary outcomes in patients. The investigators found that restrictive ventilatory defects were significantly associated with interpouch distance, duration of postoperative ventilation, the presence of gastroesophageal reflux disease, and the existence during infancy of recurrent aspiration pneumonia. The study also reported that treadmill exercise results for the patients were significantly lower than the standard population mean results. [6]
Esophageal Atresia Without Fistula
Approximately 5-8% of patients with esophageal anomalies have esophageal atresia without a fistulous connection to the trachea. Due to the long gap between upper and lower pouches, surgical correction of the deformity usually cannot be performed during the first few days of life.
Diagnosis and clinical findings
During intrauterine life, patients have polyhydramnios, and the diagnosis can be suspected with intrauterine ultrasound due to the absence of a stomach bubble. In the neonatal period, infants usually present with excessive drooling and a scaphoid abdomen. Radiologic findings pathognomonic of pure esophageal atresia include a dilated upper pouch and absence of air below the diaphragm. In 2009, Holland et al reviewed esophageal atresia cases without fistula. The most common associated anomalies were cardiac (19%), followed by renal (16%), vertebral (17%), and ano-rectal in 7% of cases. [7] The initial esophageal gap averaged 5 vertebral bodies.
Treatment
Continuous suction of the upper pouch and a feeding gastrostomy constitute initial treatment. The patient then is followed closely with contrast radiographs until primary esophagus repair can be performed, usually at an age of 2-3 months. If the gap between the proximal and distal segments of the esophagus is less than 2 vertebral bodies, the defect can be repaired with a tension-free primary anastomosis. If after an age of 3 months, the gap is still excessive, however, the esophagus can be reconstructed using a gastric or colonic graft.
Another technique consists of creating a cervical esophagostomy with traction on the distal end. The upper esophagus is elongated gradually by repeatedly mobilizing it and placing the esophagostomy lower on the anterior chest wall until a primary anastomosis can be performed.
Tracheoesophageal Fistula Without Atresia
Incidence of TE fistula without atresia varies between 1-11% of esophageal malformations.
Diagnosis and clinical findings
Patients usually present in the neonatal period. Presentation symptoms can range from coughing and choking spells (precipitated by feeding), to abdominal distention due to passage of air from the fistula, to recurrent and severe pneumonias that often are unresponsive to antibiotics. Once the diagnosis is suspected, barium swallow with cineradiography is recommended, but bronchoscopy remains the investigation of choice to confirm diagnosis.
Treatment
Most H-type fistulas are above the clavicle and can be approached through a cervical incision. Intrathoracic fistulas require a thoracotomy for adequate access. Once identified, the fistula is divided and oversewn. Preplacement of a Fogarty catheter through the fistula (by tracheoscopy) facilitates intraoperative localization of the fistula and minimizes the risk of recurrent laryngeal nerve damage. Endoscopic obliteration of the fistula has been attempted with various techniques, such as tissue adhesives, electrocautery, and sclerosants. A 2006 study of 192 patients with tracheoesophageal fistulae concludes that fistulae that have not closed after 2 endoscopic attempts are not suitable for further endoscopic treatment; therefore, an external approach should be recommended. [8]
Follow-up
Cimador et al studied the effect of postoperative morbidity during a long-term follow-up (6-12 y) in children with esophageal atresia who were treated at birth with primary anastomosis. [9] Their results demonstrated that gastroesophageal reflux (GER) and esophageal dysmotility, which are reported as frequent findings in patients who underwent primary repair, do not cause any relevant impairment to the quality of their nutritional habit.
Legrand et al concluded that the high incidence of late sequelae in esophageal atresia type III indicates the need for regular and multidisciplinary follow-up exams into adulthood. [10]
Laryngotracheoesophageal Cleft
A laryngotracheoesophageal cleft (LTEC) is a very rare anomaly with a midline defect of varying length between the posterior larynx and trachea and the anterior wall of the esophagus. Severe forms of the defect are lethal.
Classification
Evans, in 1985, classified LTEC into the following 3 categories: [11]
-
Type I (31%): Clefts are limited to the interarytenoid region above the vocal folds. This type does not involve the cricoid cartilage.
-
Type II (47%): This type includes the cricoid and extends into the cervical trachea.
-
Type III (22%): This type involves the thoracic trachea.
A modification of this classification was proposed by Benjamin and Inglis. [12] In their classification, type I cleft is limited to the supraglottic lumen above the vocal folds. Type II is a partial cleft of the cricoid extending below the level of the vocal folds, and type III involves the whole cricoid cartilage and may extend to the cervical TE septum. Type IV involves a major part of the TE wall in the thorax (see the image below).
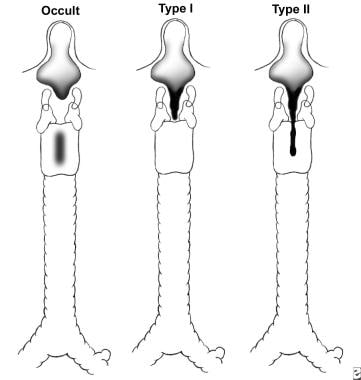
Associated anomalies
Moungthong and Holinger reported on their experience with LTEC in Chicago. [13] Cardiovascular anomalies most frequently were seen (eg, pulmonary valvular stenosis, aberrant innominate artery, patent ductus arteriosus, aortic valvular stenosis, ventricular septal defect). Pulmonary agenesis, bronchoesophageal fistula, TE fistula, rudimentary uterus, and congenital blindness also were documented.
Diagnosis and clinical findings
Type I (Benjamin and Inglis classification) or the supraglottic type is the most difficult to diagnose. Clinicians may be alerted by feeding difficulties, husky cry, or aspiration pneumonia. Stridor, coughing, and cyanotic episodes precipitated by feeding are all symptoms that may vary in severity depending on cleft extent. The differential diagnosis should include TE fistula, esophageal atresia, and choanal atresia. Chest radiographs may demonstrate pneumonia, and the lateral view may show the anterior displacement of the nasogastric tube. Definite diagnosis is made endoscopically. Suspension microlaryngoscopy is necessary to avoid missing the subtle defect (see the image below).

Management
Prevention of the gastroesophageal reflux is important in all types of clefts. Type I (Benjamin and Inglis classification) is usually corrected with growth. It requires nursing in the upright position and thickening of formula. Endoscopic repair may be needed to correct cases unresponsive to conservative measures. Types II and III can be corrected through cervical incision, while type IV requires lateral thoracotomy incision combined with a cervical approach. Others require an anterior transtracheal repair. The pleura are used for interposition in the thoracic defect, and the sternocleidomastoid muscle is used for the cervical portion. Primary closure of small defects (those with redundant mucosa) and use of costal cartilage graft interposition (for type III) have also been described. Repair of a type IV with a cardiopulmonary bypass has been described. [14]
Esophageal Stenosis and Webs
Congenital stenosis of the distal esophagus and esophageal diaphragms or webs have been reported. [15] These anomalies have been classified histologically as follows:
-
Group I - Tracheobronchial rests (cartilage, respiratory mucus glands, ciliated epithelium)
-
Group II - Membranous diaphragm
-
Group III - Fibromuscular stenosis
Diagnosis and clinical findings
Patients may present with aspiration and recurrent pneumonia, as with TE fistula. Once solid food is introduced, dysphagia and regurgitation become obvious. Endoscopy with biopsy and pH monitoring of the esophagus help to eliminate the possibility of a stricture secondary to gastroesophageal reflux. Barium swallow demonstrates narrowing of the distal esophagus.
Treatment
Pneumatic dilatation under fluoroscopy may be diagnostic and therapeutic; while it expands and cures a fibromuscular stenosis, a persistent "waist" in the balloon indicates a cartilaginous ring and the necessity for resection. Laser lysis of webs has been described and may be attempted in selected cases.
Foregut Duplications
Foregut duplications include esophageal cystic or tubular duplications and bronchogenic cysts. Due to foregut derivation of the duplications, they can be associated with an extralobar sequestration or a stenosis/atresia of the esophagus. Cysts are usually located in the right posterior mediastinum. Esophageal duplications may be separated from the esophagus or may share a common wall, but they are rarely in continuity with it. Duplications may contain gastric mucosa.
Duplications of the esophagus also can be associated with vertebral anomalies and intraspinal cysts and are often associated with intra-abdominal intestinal duplications. Most authorities ascribe these anomalies to failure of the notochord to detach from the endoderm, with persistence of a neurenteric canal.
Diagnosis and clinical findings
Large duplications can result in cardiac or respiratory symptoms. Neurologic symptoms often predominate if there are associated vertebral anomalies.
A chest radiograph may demonstrate a soft tissue mass with a mediastinal shift. Ultrasound can help distinguish a solid from a cystic mass, and a barium contrast study can demonstrate extrinsic compression of the esophagus. CT scan delineates anatomy of the mass prior to surgical resection, and nuclear (technetium) scan may identify ectopic gastric mucosa.
Treatment
Definitive treatment involves surgical resection of the duplication. Perger et al (2006) reported on 2 recent cases of thoracoscopic resection of esophageal duplication cysts. [16]
Congenital Bronchopulmonary Foregut Malformations
Congenital bronchopulmonary foregut malformations are very rare anomalies that result in a communication between the esophagus and a sequestered part of the lung. [17, 18] Symptoms include respiratory distress, cough during feeding, and recurrent pneumonias. Chest radiographs and ultrasound usually demonstrate the mass with a mediastinal shift. Barium contrast studies can demonstrate extrinsic compression of the esophagus. CT scan and/or MRI delineate pathology prior to surgical resection. Definitive treatment is surgical.
Other Rarer Anomalies
Congenital diverticulum of esophagus
Symptoms include emesis, regurgitation, and recurrent lung infections. Treatment is surgical excision of the diverticulum, fundoplication, and pyloroplasty. The video below demonstrates robotic-assisted esophagectomy pyloroplasty.
Congenital short esophagus
This condition is usually associated with a hiatus hernia and an intrathoracic stomach. Infants usually present during the first month of life with failure to thrive. Surgical intervention involves an antireflux procedure, often combined with an esophageal-lengthening procedure (eg, Collis-Nissen fundoplication).
-
Common types of esophageal atresia include (A) pure atresia of the esophagus, (B) esophageal atresia with proximal tracheoesophageal (TE) fistula, (C) esophageal atresia with distal TE fistula, and (D) esophageal atresia with double fistula.
-
The Benjamin and Inglis classification of posterior laryngeal cleft.
-
Laryngotracheoesophageal cleft, type I (Benjamin and Inglis classification).
-
Robotic-assisted esophagectomy pyloroplasty. Courtesy of Memorial Sloan-Kettering Cancer Center, featuring Inderpal S. Sarkaria, MD.





