Practice Essentials
Congenital tracheal malformations may be either disorders intrinsic to the trachea itself or may represent external forces compressing the airway.
Signs and symptoms of congenital tracheal malformations
Although congenital tracheal malformations are by definition present at birth, at times they may not cause symptoms until far later in life. The primary presenting symptom is most commonly biphasic stridor, with a considerably prolonged expiratory phase. Nevertheless, other airway-related symptoms (eg, wheezing, cough, pneumonia, croup) may be present as well. A history of frequent neck hyperextension is common as the child tries to maximally open the airway.
Workup in congenital tracheal malformations
Many tests are available to assist in the workup of congenital tracheal malformations. The initial otolaryngologic evaluation should include a fiberoptic nasopharyngolaryngoscopy to rule out any supraglottic or glottic abnormalities. Radiographic imaging is helpful, with many available modalities to consider. Neck radiography should consist of posteroanterior (PA) and lateral view high kiloelectron volt (keV) airway radiographs rather than soft tissue images. Similarly, the chest radiographs should include PA and lateral views. The yield from these studies, however, is rather low. [1]
Barium swallow studies may reveal extrinsic esophageal compression. In a child with dysphagia, a dynamic fluoroscopic study may be considered. Computed tomography (CT) scanning of the neck and chest provides additional data, while magnetic resonance imaging (MRI) together with MR angiography (MRA) can provide detailed vascular information and may obviate the need for conventional contrast angiography. However, the criterion standard diagnostic modality is still direct laryngoscopy with rigid bronchoscopy and possible rigid esophagoscopy.
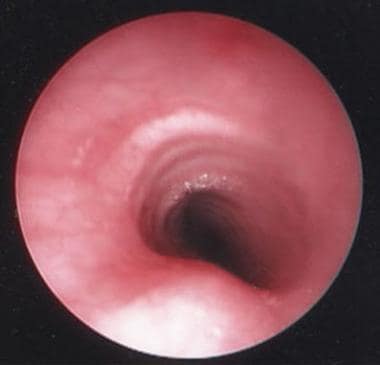
The image below depicts a healthy trachea.
Management of congenital tracheal malformations
Tracheal agenesis and atresia
Extracorporeal membrane oxygenation (ECMO) may be temporizing. Operatively, an airway conduit can be created, with eventual esophageal reconstruction. [2] Despite intervention, the mortality rate has been reported as 85% within 2 days of life. [3]
Tracheal webs and stenosis (fibrous constrictions)
Treatment for a tracheal web consists of rupturing the web. This may be accomplished via rigid dilatation through use of a laser or other ablative or cutting instruments. Only the most thickened webs require a more invasive open surgical approach; local resection is the treatment of choice.
Mild stenosis may require multiple dilatations, which may be performed either with tracheal dilators or with progressively larger rigid bronchoscopes. Short stenoses may be removed directly via a tracheal resection with primary end-to-end anastomosis. More severe lesions require tracheal reconstruction, or tracheoplasty, which generally involves placement of a graft harvested from the costal cartilage. [4]
Tracheomalacia
Symptoms of tracheomalacia are generally self-limiting because adequate growth in the diameter of the trachea eliminates symptoms by the time the patient is aged 2 years. Treatment, therefore, is supportive and aimed at preventing atelectasis, which may require administration of mucolytics to decrease secretions. For severe cases, continuous positive airway pressure (CPAP), or both may be necessary until the tracheomalacia is outgrown.
Surgical management with tracheostomy may be required in severe cases for continuous positive pressure. Internal airway stenting with endoscopic intervention and external tracheal wall support with resorbable buttress have been described.
Vascular anomalies
Because symptoms often resolve after several years, operative intervention is reserved only for cases with severe symptoms. Vascular tracheal anomalies are heterogeneous anomalies; therefore, no single surgical operation is defined. Cardiothoracic surgery is the most common service to address surgical needs. Most frequent management is with vessel pexy or division with or without re-implantation.
Complete or deformed rings
For complete tracheal rings, endoscopic techniques include laser incision and dilation and/or tracheal stenting. Open techniques of tracheal resection, tracheoplasty with pericardial or alloplastic grafting, and tracheal slide procedure for definitive management have been described. For thoracic tracheal involvement, surgery may require ECMO or temporary cardiac bypass for surgical management.
Absent or deformed tracheal cartilage rings can also occur, with resulting poor local structural support of the airway in the affected areas. Management involves watchful waiting only, because symptoms resolve with tracheal growth as the diameter of the tracheal lumen increases.
Tracheoesophageal fistulas and tracheal pouches
Surgical management is standard for tracheoesophageal fistula with atresia.
Tracheal cleft
Open surgical repairs via a transtracheal or lateral pharyngotomy approach using interposition tibial periosteum or costochondral cartilage is an option for deep type II, and III clefts. Repair of type IV clefts may also be achieved, with a transcervical approach with cricotracheal separation and reanastomosis. [5]
Tracheal Agenesis and Atresia
Tracheal agenesis and atresia are almost uniformly fatal and are fortunately quite rare, occurring in 1 out of 50,000 births. [6] The trachea may be completely absent (agenesis), or it may be partially in place but considerably underformed (atresia). In either case, communication between the larynx proximally and the alveoli of the lungs distally is lacking. Because of the lack of a normal continuous airway, affected newborns survive only if an alternate pathway for ventilation (eg, a patent bronchoesophageal fistula) exists.
Polyhydramnios is a nonspecific finding present in 70% of these patients. [6] Presentation includes respiratory distress, absent audible cry, and inability to intubate. Often, esophageal intubation is repeatedly performed. Extracorporeal membrane oxygenation (ECMO) may be temporizing. Operatively, an airway conduit can be created, with eventual esophageal reconstruction. [2] Despite intervention, the mortality rate has been reported as 85% within 2 days of life. [3]
Tracheal Webs and Stenosis (Fibrous Constrictions)
A tracheal web consists of a thin layer of tissue draped across the tracheal lumen. While the thickness of the web may vary, no deformity or abnormality of the underlying cartilage framework exists (in contrast to tracheal stenosis). The web is not complete; the degree of ventilatory symptoms present is directly related to the size of the remaining tracheal lumen. Treatment consists of rupturing the web. This may be accomplished via rigid dilatation through use of a laser or other ablative or cutting instruments. Only the most thickened webs require a more invasive open surgical approach; local resection is the treatment of choice.
In tracheal stenosis, a rather rare condition, pathological involvement continues to a much greater underlying tissue depth when compared with a web. A single tracheal ring, multiple rings, or even the entire length of the trachea may be involved. While affected patients may be symptomatic at birth, symptoms may be delayed several months until the airway lumen is compromised further by exacerbation of an upper respiratory tract infection. Symptoms include dyspnea, as well as a biphasic stridor with a prolonged expiratory component. If an affected patient requires intubation, efforts should be made to extubate early to prevent development of further edema and acute additional airway narrowing. Various other abnormalities are associated with tracheal stenosis, including vascular slings, tracheoesophageal fistulas, pulmonary hypoplasia, and trisomy 21.
The criterion standard for diagnosing tracheal stenosis is rigid bronchoscopy. [7] Other adjunct tests include CT scanning and MRI. While contrast tracheobronchography is available, its use is reserved for cases of such tight stenoses that rigid bronchoscopy is not possible even with the smallest available telescopes.
If stenosis is minimal, treatment may consist of only conservative management. [8] Mild stenosis may require multiple dilatations, which may be performed either with tracheal dilators or with progressively larger rigid bronchoscopes. While the airway may be secured via a tracheotomy if the stenosis is high in the trachea, this option is not available for more distal obstructions. Short stenoses may be removed directly via a tracheal resection with primary end-to-end anastomosis. More severe lesions require tracheal reconstruction, or tracheoplasty, which generally involves placement of a graft harvested from the costal cartilage. [4]
A study by Wang et al indicated that in cases of congenital tracheal stenosis associated with tracheal bronchus and congenital heart disease, either tracheal end-to-end anastomosis or side-slide tracheoplasty can be successfully used for stenotic correction. The study included 24 patients, with two operative deaths occurring (as a result of postoperative tracheomalacia or residual main bronchial stenosis) and with complete follow-up achieved in 19 patients. Aside from one case of tracheal granulation, patients experienced significant improvement in clinical symptoms. [9]
A study by DeMarcantonio et al indicated that compared with children suffering from tracheal stenosis alone, children with tracheal stenosis and pulmonary abnormalities, including unilateral pulmonary agenesis and hypoplasia, who undergo slide tracheoplasty tend to have a more difficult time postoperatively. Problems in study patients with pulmonary malformations included a more frequent need for prolonged mechanical ventilation and for extracorporeal membrane oxygenation and a higher mortality rate (22.2% in children with pulmonary abnormalities vs 3.6% in those without them). [10]
A study by Hosna et al indicated that in patients with inoperable tracheal stenosis, significant clinical improvement can be achieved with cryotherapy. The report included 67 patients, most of whom had tracheal stenosis or subglottic stricture, although bronchial, glottic, or thyroid isthmus stricture or stenosis was present in a minority of patients. Treatment consisted of cryotherapy alone or with balloon dilation. Improvement was experienced by 48 patients (71.6%), with another 13 (19.4%) becoming asymptomatic. [11]
Tracheomalacia
In tracheomalacia, the supporting structure of the trachea is too floppy, resulting from weakness of the tracheal walls. In addition, the posterior or membranous portion of the trachea, which does not add support to the trachea, may be wider than normal.
Patients exhibit an expiratory stridor, which may resemble wheezing. During heavy breathing, the membranous posterior wall advances anteriorly, where it may approach or even touch the cartilaginous anterior tracheal wall, thus markedly reducing the airway lumen. Symptoms are more prominent in an older infant as the respiratory movement increases. Therefore, posterior tracheal wall migration increases as well. As a result, the affected infant may hyperextend his neck in order to adequately keep his airway open.
Primary tracheomalacia is a congenital disorder of the tracheal rings and is relatively rare. On the other hand, secondary tracheomalacia is an acquired disorder in which cartilage weakness results from chronic wearing from some external pressure. Common causes of secondary tracheomalacia include vascular anomalies, such as innominate artery compression and tracheoesophageal fistula (see below). Weakness may persist or even worsen immediately following vascular repair; because the external compressing/supporting structure has been removed/repaired, the area of tracheomalacia generally scleroses and firms with time, which may be months to years.
Tracheomalacia may be diagnosed with airway fluorography. Flexible bronchoscopy is better than rigid for evaluation of the dynamic collapse of the airway during respiration. [12] Perform rigid bronchoscopy with the patient spontaneously ventilating because positive pressure ventilation from the respirator may balloon the trachea, thus hiding its floppiness.
Symptoms of tracheomalacia are generally self-limiting because adequate growth in the diameter of the trachea eliminates symptoms by the time the patient is aged 2 years. Treatment, therefore, is supportive and aimed at preventing atelectasis, which may require administration of mucolytics to decrease secretions. For severe cases, continuous positive airway pressure (CPAP), or both may be necessary until the tracheomalacia is outgrown.
Surgical management with tracheostomy may be required in severe cases for continuous positive pressure. Internal airway stenting with endoscopic intervention and external tracheal wall support with resorbable buttress have been described.
Vascular Anomalies
In tracheal disorders of vascular etiology, the trachea is inherently normal. However, large vessels are extrinsically impinging upon it, and possibly upon the esophagus as well. This is due to abnormal development of branchial arch system. Vascular anomalies are rare, and are frequently associated with other cardiac abnormalities. Abnormalities can occur such as anomalous branching pattern of the vessels originating from the aortic arch, abnormal positioning of the aortic arch itself, interrupted or supernumerary arches, or anomalous origin of the pulmonary artery from the contralateral pulmonary artery or ascending aorta.
The degree of respiratory problems and/or feeding difficulties varies depending on degree and site of compression at the trachea, the bronchi, and/or the esophagus. Symptoms range from asymptomatic to severe respiratory distress. This may present as recurrent pulmonary infection, cough, stridor, and /or dysphagia. The infant may hyperextend the neck in an effort to straighten the compressed airway, thereby stretching the trachea and pushing away the compressive extraluminal vessels. These anomalies are associated with reflex apnea and self-resolving cyanosis spells or acute life threatening events.
Diagnosis can be made with barium esophagogram, echocardiography, computed tomography (CT), magnetic resonance imaging (MRI), and angiography may aid in diagnosis. Rigid bronchoscopy and esophagoscopy allows for definitive evaluation of the airway impact and to identify synchronous tracheobronchial anomalies including complete rings and abnormal bronchial take off.
Because symptoms often resolve after several years, operative intervention is reserved only for cases with severe symptoms. Vascular tracheal anomalies are heterogeneous anomalies; therefore, no single surgical operation is defined. Cardiothoracic surgery is the most common service to address surgical needs. Most frequent management is with vessel pexy or division with or without re-implantation.
The most common vascular anomaly is compression from the innominate artery. This is likely to occur especially in cases in which the take-off of the innominate from the aorta is more distal (ie, further left) than normal. Effects are typically seen on the anterior airway only and may be observed as a pulsatile deformation of the anterior trachea of rigid bronchoscopy.
The second most common vascular anomaly is the complete vascular ring, also known as the double aortic arch. This condition provides circumferential compression of both the trachea and esophagus. Additionally, a secondary tracheal cartilage deformity (tracheomalacia) may be present from the resulting pressure. Symptoms include both dysphagia and dyspnea, and stridor may be present as well. The barium swallow study reveals circumferential narrowing. Tracheal narrowing is observed on rigid bronchoscopy, while findings on rigid esophagoscopy are remarkable for posterior esophageal compression (vs no esophageal compression with innominate artery compression). Treatment consists of surgically dividing the ring, generally by ligating the smaller of the 2 aortic arches.
A pulmonary artery sling is also known as an aberrant left pulmonary artery. In these cases, the left pulmonary artery passes between the trachea and esophagus, resulting in distal tracheal and right bronchus compression (see the video below). It is associated with the presence of complete tracheal rings. Barium swallow or rigid esophagoscopy study reveals anterior esophageal compression (in contrast to posterior esophageal compression with a double aortic arch). Treatment involves surgical rerouting of the aberrant vessel.
Complete or Deformed Rings
Abnormal development of the tracheal rings likely occurs after 8 weeks' gestation. The typical C-shaped cartilage is fused posteriorly, and the posterior membranous trachea is lacking. Incidence is estimated to be 1 in 64,500 and represents only 0.3% to 1% of all laryngotracheal stenoses. The posterior membranous portion of the trachea is absent, leading to fixed, narrowed dimension of the trachea. This may involve few tracheal rings, more than 50%, or the entire length of the trachea (sleeve trachea). [13] Complete tracheal rings have been associated with cardiac anomalies, tracheoesophageal fistulae, Down syndrome, and Pfeiffer syndrome. [6]
Symptoms of complete tracheal rings may include dyspnea, retractions, and noisy breathing often described as washing machine breathing on auscultation. Stridor may be inspiratory (cervical trachea), expiratory (thoracic trachea), or biphasic. Symptoms may not be apparent until more than 50% stenosis has occurred and may be uncovered in a setting of respiratory illness (which exacerbates the narrowing).
Diagnosis may be made with plain chest films, which may provide indication of stenosis by a narrowed air column. Airway fluoroscopy can be utilized to assess narrowing and associated pulmonary tree anomalies, which are present in up to 20%. CT or MRI along with vascular studies may be used to further evaluate the stenosis, as well as to evaluate for vascular malformations/anomalies and extrinsic compression. Rigid bronchoscopy remains the gold standard for diagnosis to assess the length of involvement.
Subclinical stenosis may be monitored, though it is important to treat associated acid reflux, and respiratory illness may require steroids and close monitoring. In addition, tracheal clearance may be affected, leading to recurrent pneumonia, which would require culture-directed antibiotic therapy.
Surgical techniques vary widely. Endoscopic techniques include laser incision and dilation and/or tracheal stenting. Open techniques of tracheal resection, tracheoplasty with pericardial or alloplastic grafting, and tracheal slide procedure for definitive management have been described (see the video below). For thoracic tracheal involvement, surgery may require ECMO or temporary cardiac bypass for surgical management. Vascular malformations (described above) are present in up to 50% of cases.
Absent or deformed tracheal cartilage rings can also occur, with resulting poor local structural support of the airway in the affected areas. Diagnosis is made by rigid bronchoscopy; flaccid segments may be observed and palpated. Management involves watchful waiting only, because symptoms resolve with tracheal growth as the diameter of the tracheal lumen increases.
Tracheoesophageal Fistulas and Tracheal Pouches
Tracheoesophageal fistulas and pouches comprise a heterogeneous group of anomalies. The etiology is likely multifactorial, with 50% associated with other malformations. The incidence is thought to be 1 in 2500 to 4500 live births. Associated conditions include VACTERL association, CHARGE syndrome, Fanconi anemia, Opitz G/BBB syndrome, and Goldenhar syndrome.
Various degrees of esophageal atresia with or without associated fistula connection to the trachea prevent egress of saliva and feed into the stomach and provide direct connection between esophagus/gastric contents with the tracheobronchial tree. The Ladd and Gross classification is used to describe the anatomy (see below). The most common presentation is proximal esophageal atresia with a distal tracheoesophageal fistula (Type C, 84%): [14]
Type A- Esophageal atresia (EA) without fistula (6%)
Type B- EA with proximal fistula (5%)
Type C- EA with distal fistula (84%)
Type D- EA with double fistula (1%)
Type E- Tracheoesophageal fistula without atresia- H-type (4%)
Respiratory symptoms are often exacerbated by associated tracheobronchomalacia. Many diagnosed prenatally, and if not most affected infants are symptomatic within first few hours of life. Symptoms include excessive saliva, pooling of secretions, feeding difficulties with coughing, regurgitation, cyanosis with feeds, and, potentially, respiratory distress.
Diagnosis may be suggested by failure to pass a nasogastric tube. A barium swallow study may reveal the fistula. A minimal amount of barium should be used because of the high risk of aspiration, and the barium should be inserted via a thin lumen tube placed past the cricopharyngeus to protect the larynx and minimize aspiration. During rigid bronchoscopy, the fistula entrance may be observed in the posterior tracheal wall; cannulation of the fistula may facilitate surgical repair. Bronchscopy additionally allows evaluation of synchronous tracheobronchial anomalies. [15]
Medical management may be initiated prior to surgical management including: antibiotics, a sump catheter for salivary and gastric diversion to prevent pneumonitis prior to surgical management, and positioning to minimize secretion burden on lungs. Surgical management is standard. The operative approach to an infant with EA depends greatly on the specific type of anomaly present and the occurrence of associated anomalies. Pediatric surgery is often the primary team for management. [14]
A study by Woo et al suggested that in the treatment of patients with EA with or without tracheoesophageal fistula, thorascopic repair produces higher rates of vocal cord paresis than does open repair, with paresis occurring in nine of the study’s 17 thoracoscopic treatment patients and in one of the 14 open surgery patients. This difference may be the result of thoracoscopic esophageal dissection high in the thoracic inlet. [16]
Other Congenital Tracheal Malformations
Tracheal agenesis and atresia are almost uniformly fatal and are fortunately quite rare. The trachea may be completely absent (agenesis), or it may be partially in place but considerably underformed (atresia). In either case, communication between the larynx proximally and the alveoli of the lungs distally is lacking. Because of the lack of a normal continuous airway, affected newborns survive only if an alternate pathway for ventilation (eg, a patent bronchoesophageal fistula) exists. Although operative techniques are available to correct the underlying abnormality, surgical attempts have yielded poor results, essentially making this a noncorrectable malformation. [14]
For patient education resources, see the Procedures Center, as well as Bronchoscopy.
Tracheal Cleft
Absence of the posterior wall of the trachea will result in a tracheal cleft. This occurs because of incomplete development of the tracheoesophageal septum. Tracheal clefts can be associated with congenital syndromes including Pallister-Hall, Opitz-Frias, and VACTERL. Laryngeal clefts are present in 6% of cases of tracheoesophageal fistulas.
The most common symptom is aspiration. As a result, an infant or child may have respiratory distress or cyanosis with feeding. Severe aspiration leads to failure to thrive or recurrent pneumonia. Excessive redundant mucosa may also cause stridor and airway obstruction. Additionally, may present with difficult intubation or difficulty ventilating secondary to large air leak.
Diagnosis is often made when a swallow study is ordered to evaluate aspiration. A modified barium swallow or fiberoptic evaluation of swallowing exam may show a posterior to anterior aspiration pattern. Formal diagnosis requires microlaryngoscopy and bronchoscopy with palpation of the posterior trachea.
Clefts are commonly described according to the Benjamin-Inglis classification. [17]
Type I: Involves the interarytenoid region down to and including the vocal cords
Type II: Extension into the cricoid cartilage
Type III: Extension through the cricoid into the cervical trachea
Type IV: Extension into the intrathoracic trachea
There is no medical management for clefts extending into the trachea. Occasionally laryngeal or cricoid clefts can be managed with thickening on liquids. Endoscopic management with suture or injection augmentation (autologous or synthetic) is available for smaller clefts.
Open surgical repairs via a transtracheal or lateral pharyngotomy approach using interposition tibial periosteum or costochondral cartilage is an option for deep type II and III clefts. Repair of type IV clefts may also be achieved, with a transcervical approach with cricotracheal separation and reanastomosis. [5] Type III and IV clefts often require ECMO or cardiopulmonary bypass (CPB) during repair. General surgery may also be involved for management of the esophagus, and a gastric exclusion procedure may be performed in large clefts. Microgastria is a commonly associated finding. Pulmonology is often involved as there is typically severe recalcitrant tracheobronchomalacia, which may lead to prolonged tracheostomy dependence.
-
Slide tracheoplasty and pulmonary artery sling repair in a patient aged 13 months. Procedure performed by Giles Peek MD, FRCS, CTh, FFICM, The Children’s Hospital at Montefiore, Bronx, NY.
-
Lateral chest radiograph shows excessive tracheal narrowing.
-
This radiograph shows the trachea during inspiration and expiration. Tracheal collapse of more than 50% during expiration is diagnostic of tracheomalacia.
-
The mechanism of tracheal narrowing is shown here in healthy cases and in cases of tracheomalacia. Adapted from Feist JH, et al. Chest 68:3, Sept, 1975.
-
Healthy trachea is visualized endoscopically.





