Embryology
General Principles of Development
Knowledge of the embryologic development of the larynx is of prime importance in understanding how congenital anomalies appear clinically and how they should be managed.
The development of the larynx can be divided into prenatal and postnatal stages.
At birth, the larynx is located high in the neck between the C1 and C4 vertebrae, allowing concurrent breathing or vocalization and deglutition.
By age 2 years, the larynx descends inferiorly; by age 6 years, it reaches the adult position between C4 and C7 vertebrae. This new position provides a greater range of phonation (because of the wider supraglottic pharynx) at the expense of losing this separation of function, ie, deglutition and breathing.
An image depicting laryngomalacia can be seen below.
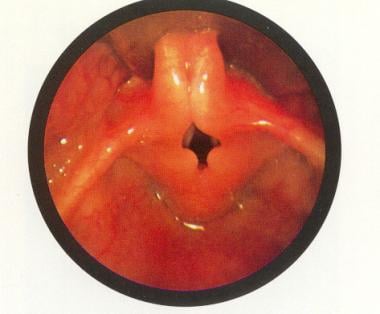 Laryngomalacia: The epiglottis is small and curled on itself (omega-shaped). Approximation of the posterior edges of the epiglottis contributes to the inspiratory obstruction. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
Laryngomalacia: The epiglottis is small and curled on itself (omega-shaped). Approximation of the posterior edges of the epiglottis contributes to the inspiratory obstruction. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
Prenatal Development
Prenatal development can be divided into the embryonic phase (0-8 wk), characterized by organogenesis, and the fetal phase, characterized by organ maturation.
Organogenesis
The larynx develops from the endodermal lining and the adjacent mesenchyme of the foregut between the fourth and sixth branchial arches. At 20 days' gestation, the foregut is first identifiable with a ventral laryngotracheal groove. At 22 days' gestation, this groove differentiates into the primitive laryngeal sulcus and the respiratory primordium; the right and left lung buds appear on day 24. The laryngotracheal groove continues to deepen until its lateral edges fuse. By day 26, this tube descends caudally, where the trachea becomes separated from the esophagus by the tracheoesophageal septum with a persistent slitlike opening into the pharynx. This fusion occurs in the caudal-to-cranial direction, and incomplete fusion results in development of persistent communication between the larynx or trachea and the esophagus. The respiratory and gastrointestinal tracts develop separately from this point.
At 32 days' gestation, the development of the larynx is first evident with the appearance of the mesenchymal-arytenoid swellings from the sixth branchial arches, just adjacent to the cranial end of the laryngotracheal tube. These swellings approximate each other in the midline and abut the caudal end of the hypobranchial eminence to convert the vertical laryngotracheal opening into a T-shaped aditus. Midline compression of the tube by these swellings results in the development of fused epithelial lamina, effectively closing the tube from the pharynx. The arytenoid swellings continue to grow cranially and to differentiate into the arytenoid and corniculate cartilages and the primitive aryepiglottic folds.
The hypobranchial eminence gives rise to the epiglottic and cuneiform cartilages, completing the supraglottic structure. The thyroid cartilage develops from bilateral chondrification centers of the fourth branchial arch, and the cricoid and tracheal cartilages develop from the sixth branchial arch. The superior laryngeal nerve emanates from the fourth branchial arch, becoming evident by day 33. By day 37, the recurrent laryngeal nerve derived from the sixth branchial arch becomes evident.
By day 40, the larynx and its cartilages and the intrinsic muscles are clearly evident. By day 44, the cartilages become more developed, and the cricoid forms a complete ring. By day 48, the epiglottis achieves its concave shape. The hyoid cartilage is observed by day 51. By the end of the embryonic phase, the larynx is clearly identifiable with its intrinsic musculature, innervation, blood supply, and cartilages. The epithelial lamina dissolves at the end of this period, resulting in a patent opening into the trachea. Continuous development of the larynx characterizes the fetal period.
Organ maturation
During the third prenatal month, the vocal processes develop from the arytenoids, and the thyroid cartilage laminae fuse in the midline. During the fourth month, goblet cells and submucosal glands become evident. The epiglottic cartilage matures to become fibrocartilaginous between the fifth and seventh months. During this period, the corniculate and cuneiform cartilages become evident. The fetal period ends with the cricoid cartilage changing from interstitial to perichondrial growth.
Postnatal Development
The main changes occurring in the larynx postnatally are a change in the axis, luminal shape, length, and proportional growth of the laryngeal elements. The larynx grows rapidly during the first 3 years of life, while the arytenoids remain approximately the same size. The arytenoids in the adult larynx are thus proportionately smaller than in the child larynx.
Beginning at age 18-24 months, the larynx descends in the neck to achieve its final position at vertebrae C4-C7 by age 6 years. During this descent, the axis of the larynx changes from a position that is slightly off horizontal (anteriorly sloped) to a horizontal position. The larynx elongates as the hyoid, thyroid, and cricoid cartilages separate from each other (accordion effect). The cricoid cartilage continues to develop during the first decade of life, gradually changing from a funnel shape to a wider adult lumen; therefore, it is no longer the narrowest portion of the upper airway.
Laryngomalacia
Epidemiology
Laryngomalacia, which is characterized by soft laryngeal cartilage, particularly in the epiglottis, is the most common congenital anomaly of the larynx, accounting for 60% of all cases. Male infants are affected twice as often as female infants. [1]
Etiology and pathogenesis
The exact cause of all cases of laryngomalacia is not known. Theories include immaturity or maldevelopment of the cartilaginous structures, gastroesophageal reflux, and immaturity of neuromuscular control.
The epiglottis is derived from the third and fourth branchial arches. Overgrowth of the third arch portion of the epiglottis results in elongation and lateral extension of the mature structure, which is observed in patients with laryngomalacia. However, histological studies do not demonstrate a difference between the quality of the cartilage structures in infants with laryngomalacia and those who have normal development.
Gastroesophageal reflux (GER) may play an etiological role in laryngomalacia. Histological evidence of gastroesophageal reflux laryngitis has recently been documented from aryepiglottoplasty specimens. The most important feature discovered was a band of inflammation of variable intensity beneath the epithelium with edema deep to it.
GER is significantly associated with severe symptoms and a complicated clinical course.
Immature neuromuscular control may be responsible for the arytenoid prolapse observed in laryngomalacia, although an increase in the incidence of laryngomalacia does not occur in premature infants who have classic hypotonicity.
Histological studies of the neuromuscular structures of the larynx do not reveal a difference between children with and without laryngomalacia.
Fetal warfarin syndrome (FWS), or warfarin embryopathy, is a rare condition that results from maternal ingestion of warfarin during pregnancy and has been associated with various congenital anomalies, including laryngomalacia.
Clinical presentation
The most common symptoms of laryngomalacia include noisy respiration and inspiratory stridor that is accentuated by being in the supine position, feeding, and agitation and that is relieved by neck extension and prone positioning. Phonation is characteristically normal. Symptoms are usually absent at birth, begin within the first few weeks of life, increase over several months, and resolve by 18-24 months of life.
Less commonly, the child may experience feeding difficulties; however, failure to thrive is rare. Respiratory distress and cyanosis are rare.
A retrospective study by Irace et al found that out of 142 pediatric patients diagnosed with laryngomalacia who presented with recurrent respiratory and/or feeding problems, 128 of them (90.1%) exhibited, via modified barium swallow study, swallowing dysfunction, and 60 of them (42.3%) displayed aspiration. The latter group included 59 patients with silent aspiration. [2]
On examination, the child with laryngomalacia is usually within the normal range of growth, appears healthy, and does not exhibit signs of respiratory distress (nasal flaring, supraclavicular or intercostal indrawing, cyanosis). The cry is normal and strong. Head and neck examination findings are usually normal. Flexible endoscopy may reveal several characteristic abnormalities, including the following:
-
Elongation and lateral extension of the epiglottis (omega shaped) that falls posteroinferiorly on inspiration (see the image below)
 Laryngomalacia: The epiglottis is small and curled on itself (omega-shaped). Approximation of the posterior edges of the epiglottis contributes to the inspiratory obstruction. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
Laryngomalacia: The epiglottis is small and curled on itself (omega-shaped). Approximation of the posterior edges of the epiglottis contributes to the inspiratory obstruction. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
-
Redundant bulky arytenoids that prolapse anteromedially on inspiration
-
Shortening of the aryepiglottic folds, which results in tethering of the arytenoids to the epiglottis
-
Inward collapse of the aryepiglottic folds (cuneiform cartilages) on inspiration
-
Expiration resulting in expulsion of these supraglottic structures with unimpeded flow of air
-
Visualization of the vocal folds, which have normal structure and function in patients with laryngomalacia
Diagnosis
The classic history and endoscopic examination usually suffice to establish a diagnosis of laryngomalacia. Radiographic studies may suggest a diagnosis of laryngomalacia and can exclude anomalies with similar presentation (eg, tracheomalacia, innominate artery compression, vascular rings of the trachea) and other causes of stridor. Inspiratory plain films of the neck may show inferior and medial displacement of the epiglottis and arytenoids. Fluoroscopy may reveal supraglottic collapse and hypopharyngeal dilatation. Rigid bronchoscopy (under general anesthesia) should be performed when the child is experiencing respiratory distress or there is suggestion of coexisting anomalies on radiologic imaging.
Laryngomalacia and obstructive sleep apnea (OSA) may co-exist. In a retrospective study, Zafereo et al (2008) documented 10 cases of laryngomalacia and OSA using polysomnography. [3]
Management
Most cases of laryngomalacia can be managed by observation, ensuring that respiratory and feeding difficulties do not develop. Medical management should include treatment of documented gastroesophageal reflux disease (GERD) because this condition is known to contribute to laryngomalacia. Treatment includes positioning measures and medications.
Surgical management is indicated in rare instances if respiratory complications develop. [4] Tracheotomy can be performed for emergent episodes of distress and should be left in place until the supraglottic pathology resolves with age. Epiglottoplasty may be performed instead of tracheotomy. In this procedure, removing a wedge of the aryepiglottic folds bilaterally, trimming the epiglottis, removing the corniculate and cuneiform cartilages, and removing the redundant arytenoid mucosa widens the laryngeal inlet. The procedure is usually performed using laser. Patients are typically extubated the day after surgery. Richter and Thompson (2008) reviewed the surgical indication, operative technique, and perioperative management of patients with laryngomalacia. [5]
Microdebrider-assisted supraglottoplasty was used by Groblewski et al (2009) to treat 28 patients. [6] It is their treatment of choice for laryngomalacia. The main outcome measures and exclusion criteria are described in their publication.
A study by Reinhard et al indicated that laser supraglottoplasty is effective in children with symptomatic laryngomalacia, finding an 86.1% overall operation-specific success rate. Factors increasing the risk of failure, however, included prematurity and the presence of type III laryngomalacia, synchronous airway lesions, and associated comorbidities. Patients had a median age at surgery of 12.7 months. [7]
A study by Cockerill et al indicated that supraglottoplasty is an effective treatment for laryngomalacia in children with Down syndrome, who are at increased risk for this condition. The study found that 89% of patients underwent successful extubation on the first postoperative day. However, the investigators reported that additional airway procedures may be needed in about half of patients. Patient age averaged 7.7 months at surgery. [8]
Complications
Laryngomalacia is generally a self-resolving condition. Complications in rare cases include chest deformities, cyanotic attacks, obstructive apnea, pulmonary hypertension, right heart failure, and failure to thrive. Importantly, synchronous airway lesions may coexist with severe laryngomalacia. Neurological conditions, hypoplastic mandible, subglottic stenosis greater than 35%, and pre-existing laryngeal edema independently adversely affect the postoperative course. [9]
Hwang et al (2013) reviewed 26 postprimary supraglottoplasty patients (for severe laryngomalacia). [10] They found that 34.6% required revision supraglottoplasty and/or tracheostomy for persistent symptoms. This persistent symptomatology was not associated with age or sex and was more commonly found in children with coexisting medical conditions. They concluded that patients with comorbidities require more than one revision supraglottoplasty.
A retrospective study by Erickson et al of pediatric patients with laryngomalacia indicated that supraglottoplasty outcomes tend to be worse in males and in patients with a neurologic diagnosis. [11]
Vocal Fold Paralysis
Epidemiology
Vocal fold paralysis is the second most common congenital anomaly of the larynx, accounting for 15-20% of all cases. No gender difference exists in the prevalence of this anomaly. [12]
Etiology and pathogenesis
Bilateral vocal fold paralysis is usually idiopathic. In certain cases, paralysis may occur secondary to central neuromuscular immaturity. Paralysis may also occur because of lesions in the central nervous system, including Arnold-Chiari malformation, cerebral palsy, hydrocephalus, myelomeningocele, spina bifida, hypoxia, or hemorrhage.
Birth trauma that causes excessive strain to the cervical spine may cause transient bilateral vocal fold paralysis lasting 6-9 months. Unilateral paralysis is usually idiopathic but may be secondary to peripheral nerve pathology. Birth trauma causing traction injuries to the recurrent laryngeal nerve may be responsible for a number of cases.
Lesions in the mediastinum, such as tumors or vascular malformations, may cause unilateral vocal fold paralysis. Iatrogenic injury to the left recurrent laryngeal nerve can occur during surgery for cardiovascular anomalies or tracheoesophageal fistulas or during neck surgery.
Clinical presentation
Bilateral vocal fold paralysis manifests as an inspiratory stridor at rest that worsens upon agitation in children with near-normal phonation and progressive airway obstruction. Obstruction can progress to a state of respiratory distress that requires airway intervention. Aspiration is common with bilateral vocal fold paralysis, often resulting in recurrent chest infections.
On examination, the child with bilateral vocal fold paralysis may or may not be in significant respiratory distress (nasal flaring, supraclavicular or intercostal indrawing, cyanosis). Head and neck examination may reveal other cranial nerve deficits. Flexible endoscopy usually elucidates the diagnosis by demonstrating vocal fold paralysis and no other abnormality.
Unilateral vocal fold paralysis may manifest during the first few weeks of life, or it may go unnoticed. The most common symptoms are a hoarse, breathy cry that is aggravated by agitation. Feeding difficulties and aspiration may also occur.
Similar to their results in patients with laryngomalacia, Irace et al found evidence that pediatric patients with unilateral vocal fold paralysis who have recurrent respiratory and/or feeding issues are at particularly high risk for dysphagia and silent aspiration. [13]
Diagnosis
A history of early inspiratory stridor associated with signs of respiratory distress suggests bilateral vocal fold paralysis. If the child is stable, flexible endoscopy may be performed to demonstrate the paralysis. Perform rigid bronchoscopy to confirm the diagnosis and to assess the airway for other anomalies. If the diagnosis is uncertain, repeat the procedure a week later to confirm the diagnosis. In patients with bilateral vocal fold paralysis, conduct radiographic and sonographic studies to assess for central nervous system abnormalities.
Flexible endoscopic examination usually suffices to establish a diagnosis of unilateral vocal cord paralysis. In the presence of respiratory distress, perform rigid bronchoscopy to exclude the possibility of concurrent airway anomalies. Conduct radiographic studies (CT scanning of the mediastinum and neck) to determine if lesions are compromising the function of the recurrent laryngeal nerve. Laryngeal electromyography (EMG) is now used in the evaluation and management of vocal fold mobility disorders and to differentiate vocal fold fixation from paralysis. It is also valuable in determining prognosis after the onset of VCP.
Management
Patients with bilateral vocal fold paralysis may need urgent airway intervention, which can usually be achieved by endotracheal intubation. Tracheotomy is necessary to relieve the obstruction and should remain in place for 2 years to allow for spontaneous recovery, which occurs completely in more than half of patients. If recovery does not occur, consider vocal cord lateralization procedures in an effort to decannulate the patient.
A study by Su et al of a simplified endoscopic suture lateralization procedure indicated that the surgery is effective in patients with bilateral vocal fold paralysis. The operation, performed in 20 patients, resulted in adequate respiration in the 19 patients who did not have an artificial airway. In addition, 19 patients had acceptable voice quality, with preoperative voice quality maintained in 14 patients. Eighteen patients suffered mild postoperative aspiration, but only for the first few days. [14]
Arytenoidectomy produces reliable results in maintaining a patent airway that supports decannulation. Transverse laser cordotomy has had early success in allowing decannulation in older children and adults.
A literature review by Thorpe and Kanotra indicated that the rate of tracheostomy is low (6.0%) in children with bilateral vocal fold paralysis who undergo glottic widening surgery, with most such patients also requiring no reoperation. The rates of tracheostomy, reoperation, and mortality were not found to differ significantly among children who were treated with cricoid split, suture lateralization, or cordectomy/cordotomy. The investigators also found that in 36.9% of cases, pediatric patients who underwent primary tracheostomy could not be decannulated. However, tracheostomized children who underwent glottic widening surgery were more likely to be decannulated than were those who were not treated with the widening operation. [15]
Supportive measures are also necessary for the patient with bilateral vocal fold paralysis to ensure that adequate nutrition is received while preventing aspiration.
Most cases of unilateral vocal fold paralysis can be managed by observation, ensuring that respiratory and feeding difficulties do not develop. Upright positioning is usually sufficient to alleviate aspiration difficulties. Rarely, intubation may be necessary to acquire a patent airway in distressed patients.
Congenital Subglottic Stenosis
Epidemiology
Congenital subglottic stenosis is the third most common congenital anomaly of the larynx, accounting for 15% of all cases. This condition is the most common laryngeal anomaly that requires tracheotomy in infants. Males are affected twice as often as females.
Etiology and pathogenesis
Incomplete recanalization of the laryngotracheal tube during the third month of gestation leads to different degrees of congenital subglottic stenosis, with complete laryngeal atresia being the extreme form (see Laryngeal Atresia). [16]
Congenital subglottic stenosis can be classified into 2 types. Membranous congenital subglottic stenosis is the result of circumferential submucosal hypertrophy with excess fibrous connective tissue and mucus glands. This type is the most common and mild form of congenital subglottic stenosis.
Cartilaginous congenital subglottic stenosis results from an abnormal shape of the cricoid cartilage. The cartilage usually narrows laterally but may also develop generalized thickening or excessively large anterior or posterior laminae.
Clinical presentation
The manifestations of congenital subglottic stenosis usually appear in the first few months of life. The stenosis is typically not evident until the child develops an acute inflammatory process, which further compromises the subglottis. The clinical presentation of a child during these periods does not differ from that of infectious laryngotracheobronchitis (croup). Biphasic stridor with or without symptoms of respiratory distress is the most common presenting symptom. The child may have a barking cough, but the cry is usually normal. Suspect congenital subglottic stenosis when these symptoms are recurrent or if they are prolonged beyond the normal duration of infectious croup (1-3 d).
Asymptomatic children who are difficult to intubate, extubate, or decannulate present another clinical scenario that arouses suspicion of congenital stenosis. Children with Down syndrome are at an increased risk of having congenital subglottic stenosis and may present in this fashion.
On examination, the child with congenital subglottic stenosis may or may not be in significant respiratory distress (nasal flaring, supraclavicular or intercostal indrawing, cyanosis). Head and neck examination findings are usually normal. Flexible endoscopy does not adequately assess the subglottis but is important to exclude the diagnoses of vocal cord paralysis and other glottic or supraglottic abnormalities. Do not pass the scope beyond the vocal cords because this may precipitate airway obstruction in a patient with a compromised subglottis.
Diagnosis
A history of recurrent croup usually suggests congenital subglottic stenosis. Perform a rigid bronchoscopy to confirm the diagnosis and to assess the airway for other anomalies. Evaluate the stenosis in terms of its length and diameter. Passing a scope or endotracheal tube through the stenosis may adequately assess the length and diameter. The largest tube or scope to pass through the airway is a good measure of the lumen diameter. Congenital subglottic stenosis is diagnosed when the lumen diameter is less than 4 mm in a term infant or less than 3 mm in a preterm infant. The findings at endoscopy that support a diagnosis of congenital subglottic stenosis are characteristically less severe than in children with acquired subglottic stenosis.
Radiographic evaluation may help assess the subglottic airway prior to bronchoscopy or when the diagnosis is unclear. Plain lateral or anteroposterior radiographs reveal a characteristic narrowing at the level of the subglottis.
Management
Most cases of congenital subglottic stenosis resolve spontaneously with growth of the child. Endotracheal intubation and tracheotomy may be required in patients who have significant airway compromise. Most children who require tracheotomy can be decannulated by age 3-4 years when the subglottic space widens.
A retrospective study by Strang et al of 132 tracheostomies performed in pediatric patients aged 12 months or younger—including in those with comorbidities such as subglottic stenosis, bronchopulmonary dysplasia, congenital heart disease, a craniofacial syndrome, or a chromosomal trisomy syndrome—found that, while the overall 12-month mortality rate was 14.4%, the mortality rate in cases with subglottic stenosis was 3.7%. [17]
Laser ablation has a limited role in the management of congenital subglottic stenosis and is usually reserved for soft lesions fewer than 5 mm in thickness. Laryngotracheoplasty is usually unnecessary but may be required to reconstruct the airway in patients who could not be decannulated. Laryngotracheoplasty is reserved for severe cases of subglottic stenosis.
Subglottic Hemangioma
Epidemiology
Subglottic hemangiomas account for 1.5% of all congenital anomalies of the larynx. Females are affected twice as often as males.
Etiology and pathogenesis
Subglottic hemangiomas develop as a result of a vascular malformation derived from the mesenchymal rests of vasoactive tissue in the subglottis.
Clinical presentation
The child is usually asymptomatic at birth. As the lesion rapidly grows from age 2-12 months, the child develops progressive respiratory distress that is initially intermittent and then continuous. Symptoms are similar to those of infectious croup, manifesting with biphasic stridor, barking cough, normal or hoarse cry, and failure to thrive. Most patients develop airway obstruction significant enough to necessitate intervention.
On examination, the child with a subglottic hemangioma may or may not be in significant respiratory distress (nasal flaring, supraclavicular or intercostal indrawing, cyanosis). Head and neck examination findings are usually normal, although half of these patients may have cutaneous hemangiomas of the head and neck. Flexible endoscopy does not demonstrate the lesion but is a necessary procedure to exclude other laryngeal anomalies.
Diagnosis
Rigid bronchoscopy is necessary to establish a diagnosis of subglottic hemangioma. The lesion is usually located posterolaterally in the submucosa in the subglottis. It may be unilateral or bilateral, or it may be located in the upper trachea. The lesion is pink-blue, sessile, and easily compressible (see the image below).
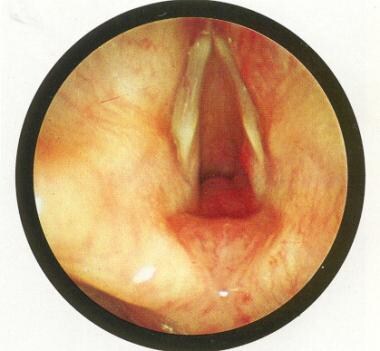 Endoscopic picture of subglottic hemangioma. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
Endoscopic picture of subglottic hemangioma. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
PHACES syndrome (posterior fossa brain malformations, hemangiomas, cardiac anomalies and coarctation of the aorta, eye anomalies with or without sternal clefts) is a neurocutaneous disorder. [18] It is important to rule out PHACES syndrome looking for subglottic hemangioma. Haggstrom et al reported recently on 17 patients younger than 1 year who were diagnosed as having large (>22 cm2) facial hemangiomas and airway hemangiomas. Eight patients (47%) met the criteria for PHACES syndrome. [19]
When the diagnosis is unclear, perform biopsy of the lesion with caution because of the risk of significant hemorrhage. Plain radiographs of the neck may show an asymmetric narrowing of the subglottis, which may aid in establishing the diagnosis prior to endoscopy.
Management
Observation usually suffices for small lesions, which do not cause significant airway obstruction. Tracheotomy is typically necessary to secure the airway until the lesion spontaneously regresses, usually by age 5 years.
Steroid injection into small- or medium-sized hemangiomas may precipitate involution secondary to suppression of estradiol stimulation of the lesion and increased responsiveness to vasoconstrictors.
Other described techniques include carbon dioxide laser ablation, cryosurgery, intralesional interferon or sclerosing agent injection, and systemic steroids. Of these techniques, endoscopic laser ablation is most frequently successful for the treatment of small unilateral lesions. The ablation of circumferential lesions in a single treatment session should be discouraged because this may lead to subglottic stenosis. External beam irradiation has a 93% success rate at curing subglottic hemangiomas but is not used today because of the increased risk of thyroid cancer. The hemangioma can recur in certain cases in which the lesion extends beyond the confines of the submucosa and goes undetected during surgery.
Open surgery has been used immediately after diagnosis as primary intervention rather than after failure of other therapies. Circular hemangioma is not mentioned as a contraindication for surgery even though a limited risk of postoperative stenosis exists in this case, especially if carbon dioxide laser treatment has already been performed.
Reports on the experience of propranolol use in the treatment of subglottic hemangiomas suggest favorable results with rapid improvement and lack of severe side effects. [20, 21, 22] Many authors suggest its use as a first-line treatment in subglottic hemangiomas when intervention is required.
Congenital Laryngeal Webs
Epidemiology
Laryngeal webs are rare congenital anomalies of the larynx.
Etiology and pathogenesis
Incomplete recanalization of the laryngotracheal tube during the third month of gestation leads to different degrees of laryngeal webs. The extreme of this situation is complete laryngeal atresia (see Laryngeal Atresia). The most common site of development of laryngeal webs is at the level of the vocal folds anteriorly, although they may occur in the posterior interarytenoid or in the subglottic or supraglottic area.
The Cohen classification of laryngeal webs separates webs into four different grades, with each varying by thickness and the percentage of glottal occlusion. For example, grade I webs tend to be thin, with less than 35% of the glottis involved, while grade IV webs are uniformly thick and involve 75-90% of the glottis.
Clinical presentation
Laryngeal webs may manifest with symptoms ranging from mild dysphonia to significant airway obstruction, depending on the size of the web. Stridor is rare except in patients who have a posterior interarytenoid web.
A third of children with laryngeal webs have associated anomalies of the respiratory tract, most commonly subglottic stenosis. Suspect other anomalies when respiratory distress is disproportionate to that caused by the web itself.
An association between anterior glottic webs and Shprintzen syndrome (also known as velocardiofacial syndrome)—chromosome 22q11.2 deletion—has been noted in a number of case reports.
On examination, the child with a laryngeal web may or may not be in significant respiratory distress (nasal flaring, supraclavicular or intercostal indrawing, cyanosis). Head and neck examination findings are usually normal. Flexible endoscopy may reveal the presence of a laryngeal web but is insufficient in evaluating the extent of this anomaly.
Diagnosis
Rigid laryngoscopy and bronchoscopy are necessary to assess the web's site, thickness, and horizontal and vertical extent. The web may appear as a thin, translucent defect involving the anterior vocal cords or as a thick, fibrous structure that extends inferiorly into the subglottis (see the image below).
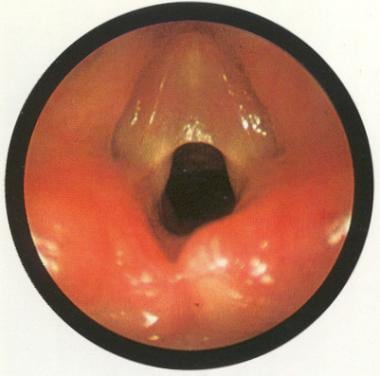 Congenital glottic web. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
Congenital glottic web. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
The posterior glottis is not usually involved but may be closed by an interarytenoid web. Closing of the posterior glottis prevents vocal cord abduction. These posterior webs are best observed when the examination is performed without an endotracheal tube in place.
Radiographic evaluation may help to assess the site and extent of the laryngeal web prior to bronchoscopy and the concurrent congenital subglottic stenosis.
Lateral plain radiographs reveal a characteristic sail sign, representing persistent tissue between the vocal cords and subglottis. [23, 24, 25]
It is also recommended that patients diagnosed with anterior glottic webs undergo fluorescence in situ hybridization evaluation for chromosome 22q11.2 deletion.
Management
Management of laryngeal webs ranges from observation to emergent tracheotomy. Thin webs may be lysed endoscopically using a cold knife or laser. Endoscopic suturing of the cut edges or placement of a keel may prevent restenosis in these cases. [26]
Thicker webs may require a laryngofissure approach, with postoperative stenting of the airway, to remove redundant soft tissue. Revision procedures may be required in certain cases, especially those that involve complicated webs.
Benmansour et al (2012) described their technique of carbon dioxide laser resection of anterior webs with the application of mitomycin C and silastic stent placement for 3 weeks. [27] They found good morphologic results, with an absence of web reformation.
A literature review by Moore et al indicated that in the treatment of less severe congenital laryngeal webs, that is, those classified as Cohen grade I or II, the rate of tracheostomy or decannulation did not differ significantly between endoscopic and open surgery. For more severe webs (ie, grades III or IV), the rate of decannulation or avoidance of tracheostomy was greater, at 96%, for patients who underwent open surgery than for those who were treated endoscopically (84%). The investigators also found that open surgery was associated with a significantly lower revision rate than were endoscopic operations (30.9% vs 77.8%). [28]
Congenital Laryngeal Atresia, Cysts, and Lymphangioma
Laryngeal Atresia
Epidemiology
Laryngeal atresia is the most rare and most devastating of the congenital anomalies of the larynx. Only a few studies report documented survivors of such lesions.
Etiology and pathogenesis
Failure of recanalization of the laryngotracheal tube during the third month of gestation leads to laryngeal atresia.
Clinical presentation
Laryngeal atresia manifests as an acute airway obstruction in the newborn immediately after clamping the umbilical cord. Examination reveals a neonate with severe respiratory distress marked by strong respiratory efforts and inability to inhale air or cry. Without immediate airway management with a tracheotomy (intubation is unsuccessful), death is imminent. The exception is a child who has a concurrent tracheoesophageal fistula of sufficient size to permit the passage of air distal to the obstruction.
Diagnosis
Diagnose laryngeal atresia endoscopically after securing the airway with a tracheotomy tube. The presence of polyhydramnios may lead to prenatal diagnosis of laryngeal atresia, allowing for management preparation prior to birth. In cases in which a tracheoesophageal fistula is present, prenatal tests are not available to help establish this diagnosis because polyhydramnios is absent.
Management
The management of laryngeal atresia involves immediate tracheotomy at birth. If the diagnosis is anticipated, avoid clamping the umbilical cord until the tracheotomy is secured to maximize oxygenation of the newborn. In patients with fistulas, mask ventilation and esophageal intubation can be used as temporary measures until a tracheotomy is performed.
Laryngeal Cysts
Epidemiology
Laryngeal cysts are uncommon congenital anomalies of the larynx. Congenital saccular cysts represent 25% of all laryngeal cysts.
Etiology and pathogenesis
Obstruction of the laryngeal saccule orifice in the ventricle leads to retention of mucus, which causes saccular cysts. Ductal cysts arise from blockage of submucosal mucus glands. These cysts can occur in the vallecula, subglottis, or vocal cords. They are common in the subglottis after prolonged intubation because of irritation and blockage of submucosal glands.
Clinical presentation
Mild symptoms, varying degrees of airway obstruction, an inaudible or muffled cry, or dysphagia can accompany laryngeal cysts.
In a small number of patients, the saccular swelling may cause an external neck mass. Endoscopy reveals a bluish pink cystic lesion behind the aryepiglottic fold (lateral cyst) or emanating from the ventricle and protruding into the laryngeal lumen (anterior cyst).
Diagnosis
Endoscopy is the procedure of choice for aiding in the diagnosis of laryngeal cysts.
Management
If emergent management of a saccular cyst is necessary, endotracheal intubation is usually possible. Needle aspiration or incision of the lesion may be performed as a temporary measure, but definitive management requires endoscopic or open complete cyst excision to prevent recurrence. Forceps or laser excision can remove ductal cysts if symptoms accompany the cysts.
Laryngeal Lymphangioma
Epidemiology
Laryngeal lymphangiomas are rare congenital anomalies of the larynx. Half the cases are diagnosed in the neonatal period, and 75% are diagnosed by age 1 year.
Etiology and pathogenesis
Lymphangiomas originate from lymphatic vessel malformations.
Clinical presentation
Individuals with laryngeal lymphangioma may be asymptomatic or may present with significant airway obstruction when the lesions attain a large size. Upper respiratory tract infections may precipitate symptoms by causing a rapid increase in the size of these lesions.
Diagnosis
Endoscopy is the procedure of choice for aiding in the diagnosis of laryngeal lymphangiomas.
Management
Tracheotomy is often necessary to establish an airway in patients with laryngeal lymphangiomas. The large size and locally invasive nature of these lesions often complicate excision. Laser ablation of these lesions is the mainstay of current therapy.
Sclerosing agents are under investigation as a possible treatment modality for laryngeal lymphangiomas.
Genetic and Central Congenital Neuromuscular Anomalies
Cri du chat syndrome
Cri du chat syndrome is a congenital condition caused by a deletion of chromosome arm 5p. The frequency of the syndrome is 1 case per 50,000 births. Approximately 1% of patients with profound retardation are diagnosed with cri du chat syndrome.
Affected individuals exhibit microcephaly, hypotonia, and cardiovascular defects. The characteristic mewing cry (cat cry) occurs only during infancy. The high-pitched stridor is the result of interarytenoid muscle paralysis in an elongated larynx with a floppy epiglottis. These patients' faces are round with hypertelorism and a broad nasal root. Cleft lip/palate is observed occasionally (see the image below). The ears are angulated posteriorly, and the hair is gray in adulthood.
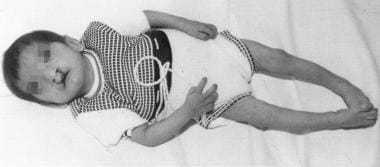 Child with cri du chat syndrome is shown. Note the cleft lip/palate and microcephaly. Used with permission from Oxford University Press [Tewfik TL, Der Kaloustian VM, eds. Congenital Anomalies of the Ear, Nose, and Throat. New York, NY: Oxford University Press; 1997.]
Child with cri du chat syndrome is shown. Note the cleft lip/palate and microcephaly. Used with permission from Oxford University Press [Tewfik TL, Der Kaloustian VM, eds. Congenital Anomalies of the Ear, Nose, and Throat. New York, NY: Oxford University Press; 1997.]
Plott syndrome
Plott syndrome is an x-linked disorder. This condition is associated with laryngeal adductor paralysis.
Arthrogryposis multiplex congenita
Arthrogryposis is associated with multiple joint disorders and central nervous system abnormalities. Laryngeal manifestations include bilateral vocal cord paralysis, hypertrophy of the cricopharyngeus, and supraglottic redundancy similar to laryngomalacia. Children with arthrogryposis multiplex congenita almost always require tracheotomy. The prognosis for individuals with this disorder is poor.
-
Laryngomalacia: The epiglottis is small and curled on itself (omega-shaped). Approximation of the posterior edges of the epiglottis contributes to the inspiratory obstruction. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
-
Endoscopic picture of subglottic hemangioma. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
-
Congenital glottic web. Used with permission from Oxford University Press [Benjamin B. Atlas of Paediatric Endoscopy: Upper Respiratory Tract and Oesphagus. New York, NY: Oxford University Press; 1981.]
-
Child with cri du chat syndrome is shown. Note the cleft lip/palate and microcephaly. Used with permission from Oxford University Press [Tewfik TL, Der Kaloustian VM, eds. Congenital Anomalies of the Ear, Nose, and Throat. New York, NY: Oxford University Press; 1997.]





