Overview
Dysmorphology is the study of disordered development resulting in recognizable morphologic abnormalities that fall outside the range of normal human variation. The late David Smith, MD, a superb dysmorphologist and the original author of the illustrated guide to syndromes entitled Recognizable Patterns of Human Malformation (WB Saunders, Philadelphia), coined the term dysmorphology. The goal of any dysmorphic assessment is to correctly interpret the pattern of structural anomalies and to arrive at the diagnosis.
The practicing specialist dealing with the ear, nose, and buccopharyngeal region is now confronted more frequently with problems related to congenital anomalies. Spectacular advances in basic and clinical genetics have brought congenital malformations and inherited disorders to the forefront of medical attention and care.
The shared spectra of dysmorphic phenotypes and their inherent variability can be overwhelming. Several thousand distinct syndromic entities have been described, and, because of their rarity, the average specialist will not have encountered the vast majority of them. Furthermore, the human genome is estimated to contain about 80,000 genes, so many rare syndromes may have yet to be defined. In addition, the popular impression of dysmorphology as an art successfully practiced only by those endowed with an unusual ability to recognize a particular gestalt may intimidate those physicians who are more comfortable with the science of medicine.
Images depicting the hands of patients with Apert syndrome can be seen below.
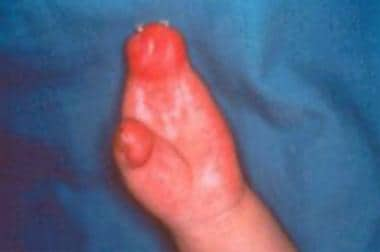
 Hand of a patient (different patient than in the image above) with Apert syndrome showing syndactyly (soft tissue).
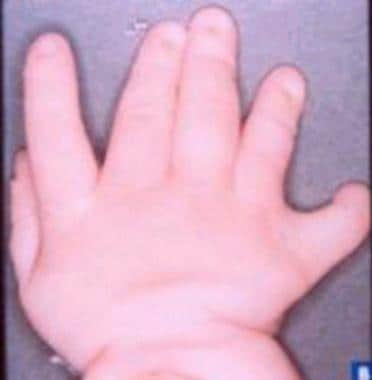
Hand of a patient (different patient than in the image above) with Apert syndrome showing syndactyly (soft tissue).
Classification
The following classification of otolaryngologic syndromes and conditions is based on system involvement. Examples of syndromes are listed with each item.
Syndromes associated with ocular disease include the following:
-
Usher
-
Norrie
-
Fraser
-
Alström
-
Bardet-Biedl
-
Cerebrooculofacial-skeletal (COFS)
Syndromes associated with craniofacial anomalies include the following:
-
Craniosynostosis - Apert, Crouzon, Pfeiffer, [1] Saethre-Chotzen, Jackson-Weiss, Carpenter, and Antley-Bixler
-
Abnormal contour - Encephalocele (with absent corpus callosum, clefting, Dandy-Walker and Arnold-Chiari malformations, ectrodactyly, and hypothalamic-pituitary disfunction)
-
Orofacial clefting - Facial clefts and associated anomalies, Tessier clefting system, lateral facial clefts, oblique facial clefts, and median mandibular defects
-
Branchial arches - Goldenhar, Treacher Collins, Nager, Miller, Wildervanck, Bixler, Möbius, and orofaciodigital syndromes (I-VIII)
-
Unusual facies - Opitz BBB, Opitz G, Noonan, Robinow, Binder, and Coffin-Siris
Syndromes associated with nervous system disease include the following:
-
Neurofibromatoses (1-8)
-
Melkersson-Rosenthal
-
Cockayne
-
Hallgren
-
Prader-Willi
Syndromes associated with integumentary system disease include the following:
-
Waardenburg
-
LEOPARD (lentigines, electrocardiographic [conduction abnormalities], ocular [hypertelorism], pulmonary [stenosis], abnormal [genitalia], retardation [of growth], and deafness)
-
Ichthyosis syndromes (Desmons; keratitis, ichthyosis, and deafness [KID])
-
Oculocutaneous albinism syndromes
-
Ehlers-Danlos
Syndromes associated with musculoskeletal anomalies include the following:
-
Contractures - Multiple pterygium, whistling face, Schwartz-Jampel, and Marden-Walker
-
Abnormal stature - Short (Aarskog, Cornelia de Lange, Hallermann-Streiff, Rubinstein-Taybi, Silver-Russell, Bloom, and Seckel), moderate (Smith-Lemli-Opitz), overgrowth (Sotos, Marshall-Smith, Weaver, and Beckwith-Wiedemann), senile appearance (progeria)
-
Osteochondrodysplasia - Achondrogenesis (types I-IV), achondroplasia, camptomelic dysplasia, chondrodysplasia punctata, Kniest, Nance-Sweeney, and osteogenesis imperfecta (types I-IV)
Other skeletal disorders include the following:
-
Craniometaphyseal dysplasia
-
Van Buchem
-
Sclerostenosis
-
Camurati-Engelmann
-
Cleidocranial dysplasia
-
Marfan
-
McCune-Albright
-
Stickler
Syndromes with urogenital anomalies include the following:
-
Branchiootorenal
-
Alport
-
Renal tubular acidosis
Syndromes with metabolic (and endocrine) anomalies include the following:
-
Mucopolysaccharidoses (I-VII)
-
Mannosidosis
-
Gangliosidoses
-
Lipidoses
-
Diabetes and associated syndromes
-
Pendred
-
Kallmann
-
Perrault
Hamartoneoplastic syndromes include the following:
-
Sturge-Webber
-
von Hippel-Lindau
-
Goltz
-
Peutz-Jeghers
-
Maffucci
Gingivodental syndromes include the following:
-
Gingival fibromatosis and its syndromes
-
Rieger
-
Trichodento-osseous
-
Llacrimoauriculodentodigital (LADD)
Syndromes associated with environmental and teratogenic agents* include the following:
-
Rubella
-
Thalidomide
-
Vitamin A
Chromosomal syndromes* include the following:
-
Trisomy 21
-
Trisomy 13
-
Trisomy 18
-
Cri-du-chat
-
Wolf-Hirschhorn
-
Turner
-
Fragile X
Associations* include the following:
-
CHARGE (coloboma, heart disease, atresia choanae, retarded growth and retarded development and/or CNS anomalies, genital hypoplasia, and ear anomalies and/or deafness)
-
MURCS (müllerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia)
-
VATER (vertebral [defects], [imperforate] anus, tracheoesophageal [fistula], and radial and renal [dysplasia]) or VACTERL (vertebral, anal, cardiac, tracheal, esophageal, renal, and limb)
Sequences* include the following:
-
DiGeorge
-
Klippel-Feil
-
Robin
-
Potter
Spectra* include the following:
Facioauriculovertebral
Oromandibular limb hypogenesis
Miscellaneous syndromes* include the following:
-
Jervell and Lange-Nielsen
-
Kartagener
-
Shprintzen
-
Coffin-Lowry
-
Kabuki make-up
-
Keutel
-
Pallister-Hall
-
Floating-Harbor
* These entries do not represent other systems; their inclusion in this classification is to aid the reader in memorizing the different conditions.
This article includes a few examples of the craniofacial syndromes, as well as 2 other syndromes frequently seen by the practicing pediatric otolaryngologist, branchiootorenal (BOR) syndrome and Waardenburg syndrome.
Apert Syndrome
Etiology
Also known as acrocephalosyndactyly type 1, Apert syndrome is inherited as an autosomal dominant trait. Increased paternal age is a factor in sporadic cases (which account for most cases). The incidence of Apert syndrome is estimated to be 15.5 cases per 1 million persons, and it represents 4.5% of all cases of craniosynostosis. Mutations have been found in the FGFR2 (fibroblast growth factor receptor 2) gene, with many cases resulting from a mutation at position 755 in which guanine is swapped for cysteine. [2] Weber et al (2010) reported on prenatal diagnosis of Apert syndrome with cloverleaf skull deformity using ultrasound, fetal magnetic resonance imaging, and genetic analysis. [3]
General manifestations
The major diagnostic criteria for Apert syndrome are craniosynostosis and syndactyly of the hands and the feet. The skull is acrocephalic with a shortened anteroposterior diameter and a flat occiput. The craniosynostosis involves the coronal suture. The forehead is prominent. The fontanelles are large and close at a later age. The palpebral fissures slant downward. The pituitary fossa and the basiocciput are significantly larger than normal. Visual impairment is a common finding; amblyopia is the major cause. Ametropia, astigmatism, anisometropia, and strabismus frequently develop in patients with Apert syndrome.
The corners of the mouth are turned down (sometimes described as a trapezoid mouth). Dental anomalies include delayed and ectopic eruptions and shovel-shaped incisors. Malocclusion with mesial molar occlusion and severe crowding of the teeth are also frequently noted.
A significant number of patients with Apert syndrome are intellectually disabled. Malformations of the corpus callosum and/or the limbic structures do occur. Other findings include gyral abnormalities, megalencephaly, and hypoplasia of the white matter and the heterotropic gray matter. Normal intelligence is present in 70% of patients.
Anomalies of the hands include syndactyly of the second, third, and fourth fingers that form a mid digital mass. The first and fifth fingers may join the hand mass or be separate from it. The distal phalanx of the thumb is often broad. Syndactyly is also noted in the feet and involves the second, third, and fourth toes. Fusion of the tarsal bones and an extra metatarsal have been noted. Other anomalies include abnormalities of the cervical vertebrae (single fusion, 37%; multiple fusion, 31%); C5 and C6 are often involved.
Moderate-to-severe acne at adolescence occurs in 70% of patients. Synostosis of the radioulnar complex, pyloric stenosis, ventricular septal defects, and polycystic kidneys are rare occurrences. Dysphagia, nutritional difficulties, and failure to thrive were reported by Pereira et al in 2009. [4]
Otolaryngologic manifestations
The nasal bridge is depressed, and the midface is typically hypoplastic. The palate is narrow and is described as Byzantine arch shaped (ie, Byzantine arch palate). Cleft palate may also occur (30%). Anomalies of the ears include otitis media, conductive hearing loss, fixation of the stapedial footplate, and wide cochlear aqueduct. Temporal bone studies revealed an absence of the internal auditory canals in one patient and enlarged subarcuate fossa in another report. Anomalies of the airway, as well as tracheal stenosis, cor pulmonale with resultant early death, were also reported. [5]
In 2010, Bannink et al reported on 11 patients treated with midface advancement for obstructive sleep apnea. Pharyngeal collapse detected on endoscopy before the surgical advancement is recommended to identify the cases that will require CPAP or tracheostomy. [6]
Mitsukawa et al (2013) performed early midfacial distractions on 11 syndromic craniosynostotic patients with obstructive sleep apnea and obtained good results. [7] The mean age of all the patients was 2 years and 5 months. Four patients in the group had Apert syndrome. They recommend this technique to reconstruct a large pharyngeal cavity, enlarge the airway, and avoid tracheostomy.
A study by Goldstein et al indicated that in patients with syndromic craniosynostosis who undergo midfacial advancement by distraction osteogenesis, complication rates vary by treatment technique. The study, which involved 54 patients (23 with Apert syndrome, 19 with Crouzon syndrome, 10 with Pfeiffer syndrome, and two with other craniofacial syndromes), found a higher rate of device displacement requiring surgery, but a lower incidence of major infections, in patients treated with halo-type distractors than in those treated with semi-buried distractor devices. [8]
Branchiootorenal Syndrome
First described in 1975, BOR syndrome (also known as Melnick-Fraser syndrome) is the association of auricular malformations, branchial fistulae, deafness, and renal anomalies. The prevalence is estimated to be 1 case per 40,000 general population.
Etiology and pathogenesis
BOR syndrome is inherited as an autosomal dominant trait with high penetrance and variable expression. The pathogenesis is presumed to be a deficiency in the differentiation of the first and second branchial arches. The anomalies of the renal system are the result of abnormal interaction between the ureteric bud and the metanephric blastema. The inner ear (stria vascularis) and the renal glomeruli share a common antigen, and more than one study group has described histopathologic changes on temporal bone examinations.
General manifestations
Renal dysplasia is reported in over two thirds of patients and varies from tapered superior poles (duplication of the collection system) to marked agenesis of the kidney. The syndrome may include Potter sequence.
Otolaryngologic manifestations
The auricular malformations include preauricular pits, which are blind pinhead-sized depressions in the superior part of the pinna of the ear. They are observed in 75-85% of patients. Other external ear malformations (40-60%) may include preauricular tags, lop or bat ears, and microtia. The external auditory canals may be atretic.
Anomalies of the middle ear include abnormalities of the ossicles, the facial nerve, and the fallopian canals. Computerized tomography of the temporal bones may demonstrate hypoplastic cochlea and absent or hypoplastic semicircular canals. Mondini dysplasia was also reported. Hearing loss (75-90%) is usually stable and may be conductive, sensorineural, or mixed. An estimated 2% of children with profound deafness have BOR syndrome.
The branchial fistulae (63%) of BOR syndrome are usually bilateral and in the lower part of the neck with external openings on the medial border of the sternomastoid muscle. Other associated manifestations include aplasia or stenosis of the lacrimal ducts (8-9%), high-arched or cleft palate, deep overbite, and facial nerve anomalies. Some authors consider hemifacial microsomia (HFM) to be a severe form. See Hemifacial microsomia, in the Goldenhar Syndrome section.
Crouzon Syndrome
Also called craniofacial dysostosis, Crouzon syndrome is characterized by cranial deformity due to premature craniosynostosis, hypoplastic midface, exophthalmos, hypertelorism, and mandibular prognathism. The incidence is estimated to be 16.5 cases per 1 million persons.
Etiology
Crouzon syndrome is inherited as an autosomal dominant trait with complete penetrance and variable expressivity. Increased paternal age is a factor in sporadic cases. Mutations have been described in the FGFR2 gene. Crouzon syndrome with acanthosis nigricans is due to a mutation in the FGFR3 gene.
General manifestations
In Crouzon syndrome, craniosynostosis of the coronal, sagittal, and lambdoid sutures may occur, giving the skull a brachycephalic shape. Occasionally, scaphocephaly or cloverleaf deformity may occur. Crouzon syndrome accounts for approximately 4.8% of general craniosynostosis cases. Proptosis secondary to shallow orbits occurs in most cases and may result in exposure keratitis. Optic nerve involvement is usually evident in 50% of cases. Other ocular findings include megalocornea, nystagmus, keratoconus, ectopia lentis, or colobomas of the iris. Frequent headaches, seizures, and mental deficiency may also occur.
A study by Driessen et al indicated that patients with a combination of Crouzon syndrome and obstructive sleep apnea tend to have earlier fusion of the spheno-occipital synchondrosis than do those with Crouzon syndrome without obstructive sleep apnea. [9]
Acanthosis nigricans has been described in association with Crouzon syndrome. Surgical treatment is usually directed toward correcting craniosynostosis, and facial abnormalities may be corrected using the Tessier procedure.
Otolaryngologic manifestations
An estimated 30-55% of patients with Crouzon syndrome experience hearing loss (usually conductive). The external auditory canals may be atretic. Ossicular fixation and deformities have also been described.
Anomalies of the palate include lateral palatal swellings in 50% of cases, but cleft lip/palate is not common. The maxillary teeth are often crowded, and an anterior open bite is usually present. The nose is often parrot-like. Calcification of the stylohyoid ligament occurs most of the time. Anomalies of the cervical vertebrae mostly involve C2 and C3.
In an analysis of nine children with Crouzon syndrome and 17 controls, using computed tomography (CT) scanning, Forte et al found the pharyngeal and nasal airway volumes of the patients with Crouzon syndrome to be comparatively reduced. The pharyngeal airway volume was 46% smaller in the Crouzon syndrome group than in controls. Moreover, there was a 31% reduction in the distance from the posterior tongue to the posterior pharyngeal wall in patients with Crouzon syndrome compared with controls. [10]
Surgical intervention for otolaryngologic manifestations involves frontoorbital and midfacial advancement, usually performed in stages during childhood. The occlusion is adequately corrected with orthodontics and final maxillary advancement after skeletal growth is completed.
Goldenhar Syndrome
Etiology and pathogenesis
Goldenhar syndrome occurs sporadically in most cases. Of reported cases, 1-2% occur in families, suggesting an autosomal dominant transmission. Animal models demonstrate that vascular disruption and hematoma formation affect the developing structures of the jaw and the ear regions in utero. Other hypotheses suggest that disturbance of the population of the neural crest cells occurs at 30-45 days' gestation. The frequency of the condition ranges between 1 case per 3500 births and 1 case per 25,000 births.
Goldenhar syndrome is also called oculoauriculovertebral (OAV) dysplasia. Gorlin, Cohen, and Levine proposed this term for the spectrum of anomalies, ranging from HFM, which denotes unilateral microtia, mandibular hypoplasia, and microstomia, to Goldenhar syndrome, which also includes epibulbar dermoids and vertebral anomalies.
General manifestations
Blepharoptosis, microphthalmia, epibulbar tumors, and retinal abnormalities have been documented in Goldenhar syndrome. Visual acuity is usually reduced. Intellectual disability varies; the frequency is 5-15% in the reported series. Children with anophthalmia/microphthalmia and cleft lip/palate seem to be at an increased risk for cerebral malformation and intellectual disability.
Congenital heart defects (CHDs) have been reported in 5-58% of the patients. Various cardiac anomalies are described. Digilio et al (2008) analyzed the frequency and anatomic features of congenital heart defects (CHD) in a series of 87 patients with OAVS. [11] CHDs were diagnosed in 28/87 (32%) patients, including 9 (32%) atrial and ventricular septal defects, 11 (39%) conotruncal defects, 4 (14%) targeted growth defects, 2 (7%) patients with situs and looping defects, one (4%) patient with a left-sided obstructive lesion, and one (4%) patient with patent ductus arteriosus. As noted in other series, the most common individual CHDs were ventricular septal defect (6 patients) and tetralogy of Fallot (classic or with pulmonary atresia, 6 patients). Renal ectopia and hydronephrosis are less common. Talipes equinovarus deformities and other limb anomalies are rare occurrences.
Otolaryngologic manifestations
In patients with Goldenhar syndrome, the mandibular ramus and condyles are hypoplastic; the maxilla, malar, and temporal bones are smaller on one side; and the mastoid is poorly pneumatized. Approximately one third of cases demonstrate bilateral involvement, and the right side is involved in 60% of cases. Macrostomia or pseudomacrostomia (lateral cleft-like extension of the corner of the mouth) usually occurs.
The parotid gland is also affected (agenesis or displaced salivary tissue). Cleft palate/lip occurs in approximately 10% of patients. Delayed dental development occurs on the hypoplastic side along with lingual and palatal malformations. Microtia or occasional anotia are present, and preauricular tags occur between the tragus and the corner of the mouth. The middle ear demonstrates ossicular anomalies and an abnormal course of the facial nerve. Hearing loss is more commonly conductive than sensorineural. Cervical vertebral anomalies occur in 30% of cases, and hemivertebrae and fused vertebrae have been described. Tracheal and pulmonary anomalies and tracheoesophageal fistula are less common.
Hemifacial microsomia
Several classifications of HFM have been proposed. The most inclusive and flexible classification is OMENS. The acronym represents each of the five major manifestations of HFM: O is for orbital distortion, M is for mandibular hypoplasia, E is for ear anomaly, N is for nerve involvement, and S is for soft tissue deficiency. The term OMENS-plus is used to include the expanded spectrum: cardiac, skeletal, pulmonary, renal, gastrointestinal, and limb anomalies.
A positive association between the severity of the mandibular hypoplasia and the severity of orbital, auricular, neural, and soft tissue involvement was demonstrated in 154 patients with HFM classified using OMENS and analyzed statistically. This study documented that HFM was associated with macrostomia (62%), facial nerve involvement (45%), ear tags (40%), palatal deviation to the normal side (39%), epibulbar dermoids (20%), horizontal discrepancy of the orbits (15%), and deformational frontal plagiocephaly (5%).
A retrospective study by Renkema et al of 991 patients with craniofacial microsomia found that 462 (47%) had extracraniofacial anomalies, including vertebral (28%), circulatory system (21%), central nervous system (11%), urogenital tract (11%), gastrointestinal tract (9%), and respiratory tract (3%) anomalies. Extracraniofacial anomalies were more prevalent in patients in whom craniofacial microsomia was bilateral, mandibular deformity was more severe, or facial nerve or soft tissue deformity existed. [12]
The management of HFM exemplifies the need for an interdisciplinary approach to craniofacial anomalies. In the past, each specialist tended to concentrate on his/her own particular area of interest or expertise. Children with HFM present not only with congenital anomalies but also with the results of abnormal growth and the potential interference of prior treatment. A systematic approach focuses on the hypoplastic mandible as the key to the management of the deformity. In infancy, treatment includes excision of preauricular tags, closure of macrostomia, and transposition of the microtic ear lobule if it is located low and anteriorly on the cheek.
Typing or grading of the mandibular deformity, as first proposed by Pruzansky and later modified, is as follows:
-
Type I is a miniature mandible with identifiable anatomy.
-
Type II is a functioning temporomandibular joint (TMJ) but with an abnormal shape and glenoid fossa. Type II is subcategorized.
Type IIA includes the glenoid fossa in an acceptable functional position (in reference to the opposite TMJ).
In type IIB, the TMJ is abnormally placed and cannot be incorporated in the surgical construction.
-
Type III indicates an absent ramus and nonexistent glenoid fossa.
The treatment concept for the child with HFM is elongation of the affected ramus (types I and II) or construction of a missing ramus and condyle (type III). Elongation or construction of the ramus creates an open bite on the affected side. This space is slowly closed over a period of 2 years with gradual reduction of an orthodontic splint. This appliance regulates eruption of maxillary teeth, thereby stimulating maxillary (midfacial) growth. The goal is to achieve a horizontal occlusal plane.
In type I HFM, a miniature mandible skeletal deformity can often be managed with a functional orthodontic appliance called an activator. This device can be placed by age 3 years. If the appliance fails to prevent obliquity of the occlusal plane, then elongation of the ramus or mandibular distraction is indicated at age 6-12 years during mixed dentition. If a type I deformity presents late in adulthood, then bilateral mandibular osteotomies are required to prevent abnormal forces from acting on the normal TMJ. A Le Fort I maxillary osteotomy may also be necessary.
Children with type II mandibles can be treated with a functional appliance; however, all children with type II HFM require mandibular elongation. Children with type IIA HFM have a well-placed, but short, ramus. They are candidates for a vertical osteotomy and elongation or a simple oblique or transverse corticotomy and gradual mandibular distraction. In children with type IIB or type III HFM, construction of the ascending ramus is needed, usually when the child has a full complement of deciduous teeth. Some have performed TMJ reconstruction in children as young as 2 years, while others have performed it in children aged 6-12 years.
Patients with type III HFM rarely present in adulthood. These patients require full construction of the mandible, the TMJ, and the zygomatic arch; in some cases, a leveling orbital osteotomy is required. A Le Fort I maxillary osteotomy is required to create a level occlusal plane and symmetry. The condyle is constructed with costal cartilage, and a bone graft is used to construct the glenoid fossa. In combination, a sliding genioplasty is often necessary, with or without a mandibular procedure as the final surgical step for the young adult with HFM.
Ear construction must be conceptualized prior to mandibular correction to avoid the improper placement of incisions and scarring. In high-grade mandibular deformity (ie, type IIB, type III), deferring ear construction until skeletal symmetry is achieved (usually after age 10 y) is recommended. In low-grade mandibular hypoplasia, the ear construction can be performed after age 6 years, when the costochondral elements are large enough to build an autogenous auricular framework.
Soft tissues can be hypoplastic in HFM. In rare cases, after restoration of the skeletal framework, soft tissue must be added. Onlay bone grafts to the ramus usually give symmetric lateral facial contour. Free tissue transfer by microsurgical techniques is another option. In minor cases, a dermal graft can be sufficient.
Mandibular distraction is a new method of ramus elongation for patients with type I and type IIA mandibular deformities; it can also be used for some patients with type IIB mandibular deformity. Ilizarov of Russia originally described this technique for long bones. An osteotomy (corticotomy) is performed, and metallic pins are placed through the hypoplastic ramus on each side of the osteotomy. The distraction device is attached and secured to the pins, and the device is turned 1 mm per day. The mean amount of lengthening (or distraction) is 24.5 mm at a rate of 1 mm per day. Mandibular distraction can be performed in children as young as 2 years.
Distraction osteogenesis (DO) gives more mandibular bone than elongation by sliding vertical osteotomy or rib graft construction. Careful follow-up documentation of the ramus height and the occlusal plane is necessary. Nevertheless, the concept and possibilities for skeletal distraction are promising for the ramus and for correction of other craniofacial skeletal abnormalities. The effect of mandibular distraction osteogenesis (DO) on developing molars should be observed. However, Kleine-Hakala et al (2007) concluded in their study that dental injuries are a minor disadvantage compared with the vast benefits offered by DO, focusing on these drawbacks might lead to reconsideration of the type of the device as well as the timing of DO. [13]
Treacher Collins Syndrome
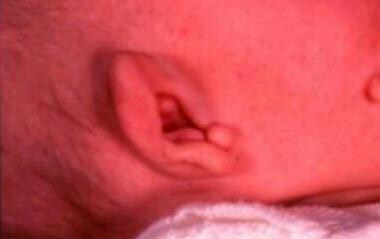
In 1900, Treacher Collins described the essential traits of the syndrome that bears his name. Treacher Collins syndrome is also known as Franceschetti-Zwahlen-Klein syndrome or mandibulofacial dysostosis. Diagnostic criteria include microtia and malformed ears (as shown below), hypoplastic midface, downward slanting of the palpebral fissures, coloboma of the outer third of the lower eyelids, and micrognathia.
Etiology
Treacher Collins syndrome is inherited as an autosomal dominant trait with variable expressivity and high penetrance. A recessive form has also been described. Experimental embryological studies using vitamin A and isotretinoin produced similar changes in rats, hamsters, and mice. Hypersensitivity of the mother to vitamin A has also been suggested. The proposed gene location is band 5q11.
Vincent et al (2013) reported on 2 patients with Treacher Collins syndrome, but with associated intellectual disability, due to a large deletion encompassing several genes, including the TCOF1 gene. [14] They mention also the involvement of deleted genes CAMK2A or SLC6A7 in the cognitive delay of the reported patients. They propose the systematic investigation for 5q32 deletion when intellectual disability is associated with mandibulofacial dysostosis.
General manifestations
Most patients have normal intelligence. Developmental retardation is reported in only 5% of cases. Delays are probably secondary to undetected hearing loss. The most common ocular findings are lower lid colobomas (77%) with absence of cilia medial to the coloboma. Microphthalmia and coloboma of the iris may be observed. Other uncommon anomalies include congenital heart defects and cryptorchidism.
Otolaryngologic manifestations
The facies are characteristic with flat supraorbital ridges; a nose that appears large because of depressed malar bones; underdeveloped lower nasal cartilages; and, often, a high bridge of the nose. Auricles are malformed, and microtia is reported in 60-77% of cases. External auditory canals are stenosed or absent. In the middle ear, varying anomalies, such as monopodal stapes, ankylosed footplate, bony bridge to the fallopian canal, and malformed incus, are described. The tympanum may be filled with connective tissue, and the epitympanum and the mastoid may demonstrate different degrees of hypoplasia or agenesis. The cochlea and the vestibule may demonstrate abnormalities.
Hearing loss is usually conductive, but a sensorineural component has also been described. One third of patients with Treacher Collins syndrome have a cleft palate or velopharyngeal insufficiency. Macrostomia and hypoplasia of the parotid glands are also observed. The coronoid process of the mandible is flat, and the condyles may be malformed. Blind fistula between the auricle and the angle of the mouth, choanal atresia, and pharyngeal hypoplasia are occasional abnormalities.
Franceschetti described 5 categories that are dependent on the number of features present. In Tessier's classification of craniofacial clefts, the complete form of the syndrome manifests craniofacial clefts 6, 7, and 8.
These patients have a complex craniofacial malformation involving the middle and lower parts of the face and affecting both bony structures and soft tissues. The deformity is always bilateral and nearly always symmetric. They have downward-sloping palpebral fissures, depressed cheekbones, deformed pinnas, receding chin, and wide oral commissures (macrostomia). This syndrome can be mistaken for bilateral HFM. The degree of symmetry and normal facial nerve function are important distinguishing characteristics of Treacher Collins syndrome.
Plain radiographs reveal the malar hypoplasia, teardrop-shaped orbits, and partial or complete absence of the zygomatic arches. In severe cases, the lateral wall and the floor of the orbits are absent, that is, a clefting of the inferolateral orbit extending into the sphenomaxillary (inferior orbital) fissure. Mandibular retrognathism is present, with shortening of the body and the ramus of the mandible. The chin is retruded severely, and the nasomaxillary area is relatively prominent.
Upper airway narrowing and hearing impairment are major issues during infancy. These infants are prone to obstructive sleep apnea and sudden infant death syndrome. The size of the nasopharynx in Treacher Collins syndrome is about 50% smaller than normal.
In a study of 1114 patients with Treacher Collins syndrome matched against a non-syndromic cohort, Kloos and Kaelber found that the risk of diagnosed otitis media was 13 times higher in the presence of the syndrome. Moreover, 40% of the patients experienced conductive hearing loss, and almost 10% suffered eustachian tube dysfunction. The investigators suggested that the eustachian tube dysfunction may have been associated with the increased otitis media risk; obstruction could have caused negative pressure to build in the middle ear, resulting in a backflow of nasopharyngeal bacteria into that space. [15]
A study by Ma et al indicated that in persons with Treacher Collins syndrome, upper airway volume is particularly diminished in the retroglossal region. The study, which included 30 patients with Treacher Collins syndrome and 35 controls, also found a correlation in the syndrome between airway volume and maxillomandibular dysmorphologies (and the relationship of these dysmorphologies to the cranial base). [16]
The surgical plan for Treacher Collins syndrome must be tailored to the patient's specific deformities, taking into consideration facial growth and psychosocial impact. The surgical correction can be separated into procedures for the upper face and for the jaws or the lower face.
Waardenburg Syndrome
Van der Hoeve and Mende described certain aspects of this syndrome, but Waardenburg first defined it in 1951.
Etiology
Waardenburg syndrome is inherited as an autosomal dominant trait with variable penetrance. Three percent of all children who are congenitally deaf have Waardenburg syndrome. Its incidence is estimated to be between 1 case per 20,000 population and 1 case per 40,000 population.
Waardenburg syndrome type 1 (WS1) and Waardenburg syndrome type 3 (WS3) have been mapped to a distal portion of band 2q3 and found to be associated with mutations in the PAX3 gene. They are considered to be allelic. Waardenburg syndrome type 2 (WS2) is heterogeneous, with about 20% of cases caused by mutations in the human microphthalmia (MITF) gene. Individuals with auditory, pigmentary, or neural crest syndromes who do not fit into the stringent definitions of Waardenburg syndrome are unlikely to have mutations in either the PAX3 gene or the MITF gene.
General manifestations
Along with congenital sensorineural deafness, multiple congenital anomalies are associated with this syndrome. The widely spaced medial canthi (dystopia canthorum) are characteristic of WS1. Lack of penetrance of this sign occurs in no more than 1% of cases. Confluent eyebrows with hypertrichosis of the medial portions (synophrys) are present in 85% of patients with WS1 and in 25% of patients with WS2.
White forelock (poliosis) occurs in 20-40% of patients with WS1 or WS2, and heterochromia irides occur in 30% of patients with both types.
Other findings include craniosynostosis, blepharophimosis, glaucoma, mild prognathic mandible, Hirschsprung megacolon, premature graying of hair, anophthalmia with limb malformations, ventriculoseptal defect (VSD), meningocele, spina bifida, hypoplasia of the shoulder girdle, and axillary webbing. Bilateral renal anomaly has also been described.
Clinical types
See the list below:
-
Type 1: Dystopia canthorum is present. Type 1 is 20 times more common than type 2.
-
Type 2: Dystopia canthorum is not present. Hearing loss is more common (50%) in this type as are pigmentary disorders.
-
Type 3: In Klein-Waardenburg syndrome or pseudo-Waardenburg syndrome, dystopia canthorum is not present, but one-sided ptosis of the upper eyelid and upper limb anomalies are present.
-
A Waardenburg-Hirschsprung disease association clinical type has been described. A novel mutation of the endothelin-B receptor gene (EDNRB) in Waardenburg-Hirschsprung disease has recently been described.
Otolaryngologic manifestations
In Waardenburg syndrome, deafness, broad nasal bridge, hypoplastic alae nasi, high-arched or cleft palate, abnormal vestibular function, and hypoplastic ear cartilage are present. Sensorineural hearing loss is either unilateral (15%, WS1; 5%, WS2) or, more commonly, bilateral (20%, WS1; 55%, WS2). The loss is usually in the low and mid frequencies and normal hearing for 6000-8000 Hz. Abnormal vestibular functions are noted in WS2 but not in WS1. Temporal bone studies described the absence of the organ of Corti, with thickening of the basal membrane and degeneration of the spiral ganglion. However, children with Waardenburg syndrome (WS) derive significant benefit from cochlear implantation (CI); this results in improvements that are comparable with implanted children in general.
Migirov et al reported on a small cohort of 5 patients from Israel. [17] All children were found to have a patent cochlea, and the electrodes were implanted into the scala tympani without difficulty. The follow-up ranged from 1.3-10.2 years of implant use. All children achieved open-set recognition of 2-syllable words, with an average score of 81%. Four of the 5 children achieved open-set recognition of monosyllabic words (average score 40%), and 3 achieved open-set recognition of words in sentences (average score 81%).
-
Hand of a patient with Apert syndrome showing syndactyly (bone and soft tissue).
-
Hand of a patient (different patient than in the image above) with Apert syndrome showing syndactyly (soft tissue).
-
Preauricular tags on a patient with Treacher Collins syndrome.





