Practice Essentials
A large proportion of the infants born deaf each year have a hereditary disorder, with permanent hearing loss having been identified early in almost 6000 US infants born in 2019. [1] Hereditary disorders must be differentiated from acquired hearing losses. Not all hereditary hearing loss is present at birth; some children inherit the tendency to develop hearing loss later in life.
Genetic sensorineural hearing loss (SNHL) includes a broad range of disorders that affect infants, children, and adults. Affected individuals may have unilateral or bilateral hearing loss ranging from mild to profound. This article, like most related discussions, focuses on childhood hearing loss, with consideration of a few forms of adult-onset hearing loss.
Signs and symptoms of hearing loss
Findings associated with hearing loss include the following:
-
Microtia or atresia of the ear canal
-
Cleft lip or palate
-
Craniofacial abnormalities - Such as micrognathia, facial asymmetry, microcephaly, or craniosynostosis
-
Cranial nerve weakness
-
Heterochromia of the iris or other abnormalities of the ocular structures
-
Vision impairment
-
Goiter
-
Skeletal abnormalities.
Workup in genetic sensorineural hearing loss
Laboratory studies
Lab studies in the assessment of genetic SNHL can include the following:
-
Molecular genetic testing [2]
-
Complete blood count (CBC) with differential
-
Chemistries
-
Blood sugar determination
-
Blood urea nitrogen (BUN) and creatinine measurement
-
Thyroid studies
-
Urinalysis
-
Fluorescent treponemal antibody absorption (FTA-ABS) test
-
Specific immunoglobulin M (IgM) assays for toxoplasmosis, rubella, cytomegalovirus (CMV) infection
-
Herpes virus autoimmune panel
-
Autoimmune profile
Imaging studies
Computed tomography (CT) scanning assists in the diagnosis of suspected labyrinthine anomalies, such as a large vestibular aqueduct or Mondini dysplasia. It may also help in identifying the relatively nondysplastic and presumably somewhat-hearing ear when auditory habilitation is being considered.
Magnetic resonance imaging (MRI) with gadolinium enhancement is the criterion standard for evaluating potential retrocochlear pathology as a cause of hearing loss. Highly T2-weighted images obtained with appropriate sagittal sections can depict aplasia of the cochlear nerve and subtle malformations of the inner ear.
Other tests
Other tests in the workup of genetic SNHL include the following:
-
Auditory brainstem response (ABR) - This is most clinically useful for assessment of infants and young children
-
Audiometry - Valid and reliable techniques are presently available to provide information relevant to presence, degree, and nature of hearing impairment in children within the first 24 hours of life
-
Otoacoustic emissions (OAEs) - OAEs are samples of measurable acoustic energy generated by vibratory patterns in the normal cochlea and propagated into the external auditory canal (EAC) by way of the middle ear apparatus
-
Electrocardiography (ECG): Consider ECG to detect cardiac conduction anomalies, especially in any child who has a family history of sudden infant death syndrome (SIDS), syncope, cardiac dysrhythmia, or sudden death in a child
Management of genetic sensorineural hearing loss
Treat any middle ear disease, including otitis media, with appropriate medical therapy.
Hearing amplification, whether with conventional or advanced technologic devices, is critical to the habilitation process. Also, assistive listening devices and personal systems may be helpful.
Consider cochlear implantation for patients who do not demonstrate significant benefit from conventional hearing amplification. Cochlear implants are electronic devices designed to convert mechanical sound energy into electric signals that can be delivered to the cochlear nerve.
Pathophysiology
Volumes of texts and journals are dedicated to the pathophysiology of genetic hearing loss and can not be easily summarized in a few paragraphs. Interestingly, note that as our understanding of the molecular basis of genetic hearing loss increases, so does our understanding of the molecular basis of hearing itself, although it remains still largely unsolved. [3, 4]
First, we must understand that genetic hearing loss seems to breach all categories of hearing loss, including the following: congenital, progressive, and adult onset; conductive, sensory, and neural; syndromic and nonsyndromic; high-frequency, low-frequency, or mixed frequency; and mild or profound. Genetic hearing loss may show patterns of recessive, dominant, or sex-linked inheritance and may be a result in mutation of both cellular or mitochondrial DNA (and RNA, in the case of mitochondrial genes). Genetic hearing loss may be subject to environment and aging, such as noise-induced or age-induced hearing loss.
New genetic mutations are linked to hearing loss every year. More than 100 loci have been identified involving genes that code for proteins involved in the structure and function of hair cells, supporting cells, spiral ligament, stria vascularis, basilar membrane, spiral ganglion cells, auditory nerve, and virtually every structural element of the inner ear. [5]
See the image below.
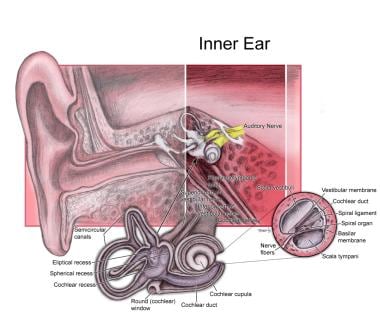 Inner ear.
Inner ear.
Dysfunctional proteins have been identified in the impaired molecular-physiologic processes of potassium and calcium homeostasis, [6] apoptotic signaling, [7] stereocilia linkage, [8] mechanicoelectric transduction, electromotility, and other processes. [3] Eisen and Ryugo provide an excellent review of the molecular pathophysiology of genetic hearing loss. [3]
Epidemiology
Frequency
United States
Between 2011 and 2012, the overall annual prevalence of hearing loss in the United States was 16% (27.7 million) among adults aged 20-69 years, a slight decrease from the annual prevalence of 16% (28.0 million) between 1999 and 2004. Moreover, at birth approximately 2-3 children per 1000 in the United States have detectable hearing loss. [9]
Congenital hereditary hearing loss must be differentiated from acquired hearing loss. More than half of all cases of prelingual deafness are genetic. The remaining 40-50% of all cases of congenital hearing loss are due to nongenetic effects, such as prematurity, postnatal infections, ototoxic drugs, or maternal infection (with cytomegalovirus [CMV] or rubella). Most cases of genetic hearing loss are autosomal recessive and nonsyndromic. Hearing loss that results from abnormalities in connexin 26 and connexin 30 proteins likely account for 50% of cases of autosomal recessive nonsyndromic deafness in American children.
The incidence of hearing loss increases with age. In the United States, hearing loss exists in about one in three people between the ages of 65 and 74 years and in almost 50% of those older than 75 years. [10] Adult-onset hearing loss can be attributed to normal aging processes and environmental triggers. However, an individual's genetic predisposition should not be underestimated, as illustrated by aminoglycoside-induced ototoxicity and the predisposition to noise-induced hearing loss.
Current statistics can be found on the Early Hearing Detection & Intervention (EHDI) Program Web site published by the Centers for Disease Control and Prevention.
International
More than 1.5 billion people worldwide suffer from hearing loss, according to the World Health Organization (WHO). [11] Genetic sensorineural hearing loss (SNHL) appears to occur twice as often in developed countries as in underdeveloped countries. In addition to ancestry and race, the proportions of hereditary versus acquired and syndromic versus nonsyndromic hearing loss across populations is highly variable and is heavily influenced by multiple factors, some likely not yet identified, including drift of populations, frequency of consanguinity, and health status.
Estimating the prevalence of hereditary hearing loss in populations across the world is very difficult because access to health care, poor health conditions, and a low level of awareness of hearing loss is compounded by a higher frequency of complicating risk factors such as neonatal distress, prematurity, high fever, otitis media, meningitis, ototoxic medications, and illnesses such as rubella. [12]
Saunders et al demonstrated a prevalence of significant hearing loss of 18% in a group of school-aged children in rural Nicaragua, with a familial history of hearing loss in 24% of the children with hearing loss. [12] Large-scale epidemiologic studies are needed and will become more feasible as molecular testing is made available to the world’s populations.
Race
Genetic hearing loss does have significant ethnic links. Angeli recently reviewed the ethnic variability of DGNB1 and showed greater allelic variability in Hispanics. [13] Schimmenti et al showed a lower prevalence of connexin-related hearing loss in Hispanic infants. [14]
Age
Before universal hearing screening for newborns, less than 50% of children who had hearing impairment were identified before the age of 3 years. Detection of risk factors (eg, prematurity, low birth weight, low Apgar scores) helps in identifying less than 50% of infants who have or who are at risk for hearing loss. In one study, 78% of infants identified with hearing loss were in the well-baby nursery and not the neonatal intensive care nursery. [15] This finding emphasized the ineffectiveness of screening on the basis of risk identification alone. Neonatal hearing screening is required in at least 45 states, resulting in earlier identification and treatment of hearing impairment. [16] Hereditary hearing loss may also be progressive or adult in onset.
-
Inner ear.





