Practice Essentials
In congenital aural atresia (CAA), the external auditory canal (EAC) and structures in the middle ear fail to develop completely. Embryologic development of the ear canal and middle ear may be arrested at any point in the process. Therefore, the clinician may encounter varying degrees of severity of this malformation. In the severe form of the disorder, no identifiable ear canal exists (complete atresia), and the middle ear and its structures (ossicles, or ear bones) may be absent or show significant underdevelopment. If a semblance of an external auditory meatus (outer opening) is present, the ear canal may end in a shallow blind pouch. In less severe forms of the disorder, the ear canal may be stenotic (narrow) with a pinpoint aperture leading into the medial ear canal and, possibly, a rudimentary tympanic membrane. The tympanic membrane in these ears may or may not be attached to the ear bones (the ossicular chain).
Surgery to repair CAA is one of the most challenging operations the ear specialist faces. The goals of surgery are to provide the patient with a clean, dry, skin-lined external auditory canal with long-term restoration or improvement in hearing.
CAA is commonly accompanied by microtia ("small ear"), or incomplete development of the auricle (pinna), which is also a surgical reconstructive challenge. [1] Microtia is classically graded I, II, and III: I is simply a smaller than normal ear with all of the normal ridges and valleys of the delicate cartilage. II is smaller than normal, with not all of the ridges and valleys of the cartilage developed. Grade III microtia is a "peanut" ear, with a small cartilage or soft tissue remnant. The term anotia refers to the complete absence of an auricle or of any ear cartilage or skin appendage.
The ear canal and auricle assume separate developmental and embryologic origins, yet the development of the auricle often mirrors that of the middle ear structures, with a more developed auricle generally being accompanied by a more developed middle ear space and ossicular chain. [2] Reconstructive options for microtia are another topic entirely and are addressed in a separate section (see also the topic Microtia).
An image depicting grade III microtia with aural atresia can be seen below.
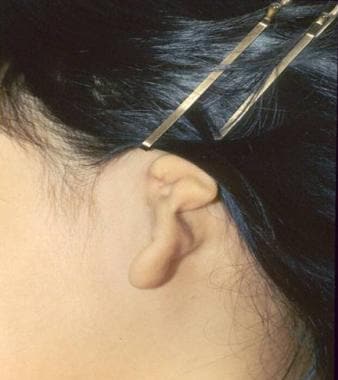 Grade III microtia with complete absence (atresia) of the external auditory canal.
Grade III microtia with complete absence (atresia) of the external auditory canal.
Evaluation and management of congenital aural atresia
Our current understanding of the evaluation and management of patients with CAA stems from 3 significant advancements. First, detailed anatomic studies of the temporal bone have improved our knowledge of the sequence of events in the development of the different regions of the ear (ie, external ear, middle ear, and inner ear). Findings from these studies explain why some regions of the ear are more developed than others in patients with CAA and enhance our appreciation of the anatomical variations found in CAA. In general, patients with aural atresia have normal inner ear (cochlear) function because the cochlea and inner ear structures develop from a separate embryological system from the ear canal and middle ear.
The cochlea and balance system (inner ear) develop from the primitive otocyst of neuroectodermal origin, while the ear canal and middle ear develop from the branchial apparatus and set of swellings and grooves on the side of the embryo's head and pouches on the inside. The implication of this difference in embryologic origin between the inner ear (cochlea) and the middle and outer ear is that patients with aural atresia, in general, have normal inner ear function. The hearing loss, therefore, is conductivein nature, meaning that there is a failure of sound energy to be conducted efficiently to the inner ear. This can be confirmed by specialized audiometric testing.
Second, high-resolution computed tomography (CT) scanning has allowed the otologist to "see" into the middle and inner ears to understand the internal anatomy. Before CT scanning, plain radiography and tomography were available, and surgeons could only surmise what the anatomy looked like inside without the ability to predict good surgical candidates. High-resolution CT scanning is paramount in the evaluation of patients for ear canal surgery (see below). With recent reports of the long-term effects of even small radiation dosages from CT scans in young children, CT scanning is typically not recommended until the child is being considered for surgery, around age 5-6 years.
Third, individuals such as Jahrsdoerfer, De la Cruz, and Lambert have gained vast experience in managing patients with CAA and have shared their insight into surgical candidacy, surgical technique, hearing restoration, and management of complications.
In CAA, the role of the otologist extends beyond diagnosis and surgical treatment. The otologist must consider the hearing needs, along with the speech and language development, of the child, especially in children with CAA in both (bilateral) ears, and must determine not only the appropriate treatment, but also the timing of that treatment. The otologist must work closely with the facial plastic surgeon to determine the optimal time for microtia and atresia repair. [3] This decision-making process usually occurs in the setting of anxious parents seeking answers as to how this deformity came about, what can be done to restore their child's hearing and appearance, and how soon treatment can be administered.
An important point to remember: While almost all patients with microtia are candidates for reconstruction, not all patients with CAA are candidates for canal surgery (atresia repair). Among patients with associated syndromes such as Treacher-Collins or hemifacial microsomia (HFM)/Goldenhar, approximately 50% are not surgical candidates, because of the existing anatomy. In patients with isolated unilateral (one-sided) CAA (most patients), approximately 65-75% are surgical candidates.
For surgical candidates with favorable anatomy, the otologist must rely on the skills, techniques, and experience from middle ear and mastoid surgery to optimize the surgical result. Surgical success is based on restoration of useful hearing, long-term stability of hearing, and maintenance of a patent, skin-lined ear canal. Otologists agree that when these goals are achieved, few accomplishments are as gratifying as successfully treating a patient with CAA.
Alternatives to surgical correction include amplification devices, such as a bone-conducting hearing aid worn on a headband or the osseointegrated bone-anchored hearing devices (eg, Ponto System [Oticon Medical, Somerset, NJ; BAHA System [Cochlear Corp., Englewood, CO]). [4, 5] Conventional hearing aids in patients with canal stenosis may also be used if the hearing device is able to provide adequate amplification. [6]
History of the Procedure
Although atresia of the external auditory canal has been recognized for over 70 years, reports of surgical repair of atresia did not surface until the late 1940s and 1950s. Nager advocated tailoring the surgical technique to open the ear canal and restore hearing to the severity of the atresia. [7] For minor malformations (group I; normal or stenotic canal with hypoplastic middle ear and some malformation of the ossicles), Nager described an endaural approach to widen the stenotic ear canal and address any middle ear abnormalities.
For group II malformations (fistulous tract or complete atresia of the canal with a bony atretic plate and some degree of malformation of the middle ear structures), Nager recommended opening the mastoid to expose the lateral ossicular mass, freeing the ossicular chain, and using a split-thickness skin graft to line the mucoperiosteal membrane on the undersurface of the bony atretic plate.
For more severe malformations (group III; complete ear canal atresia with nonpneumatized mastoid and middle ear), he advised against surgery, or possibly creating a window into the lateral semicircular canal. [7]
Schuknecht also divided aural atresia patients into 3 groups based on severity and reviewed 3 methods for surgical reconstruction: 1) creating a window into the lateral semicircular canal, 2) canaloplasty, and 3) type III tympanoplasty. [8] In all of these, the mastoid cavity is opened to access the ossicles and middle ear space, leaving the patient at risk for postoperative drainage and other problems associated with a cavity.
Jahrsdoerfer first described the anterior approach, avoiding opening the mastoid air cells, in 1978. [9] This approach, the standard in atresia surgery today, keeps the canal drilling anterior and superior by following the bone separating the ear canal from the brain and the jaw (temporomandibular) joint through the nonpneumatized bone of the atretic plate directly into the epitympanum and middle ear space. The atretic bone is carefully picked away, the ossicles are freed, and temporalis fascia is used as an eardrum graft with preservation of the native ossicular chain. Placement of a split-thickness skin graft and opening the meatus complete the procedure. With a few minor modifications, this technique is used today and continues to deliver significantly improved hearing results without a mastoid cavity and with fewer complications.
Problem
The main anatomic deformity in CAA is failure of the external ear canal to open and complete its development. The severity of the atresia is variable. The anatomy of the ear depends on the point along the embryological pathway where development is arrested. In the most severe form of CAA (ie, when development of the ear is halted very early), the ear canal cannot be identified. Bone fills or blocks the region the ear canal normally occupies, and no external opening (meatus) is present. In mild cases, the ear canal is present, but it is stenotic and markedly narrowed. A rudimentary tympanic membrane may be present, and it may or may not be connected to the ossicular chain.
In rare cases of ear canal stenosis (narrowing), skin that normally lines the ear canal can become trapped and cannot self-clean. This trapped skin causes bony remodeling of the surrounding bone and can cause infection. This trapping of skin is termed cholesteatoma and is a clear indication for surgical intervention to remove and clean out the trapped skin and provide for a safe ear that will not trap skin. Cholesteatoma should be suspected when there is moisture or drainage at the pinpoint opening of a stenotic canal. It is more definitively diagnosed by high-resolution CT scan of the temporal bone (see "Clinical picture with canal stenosis," below). Magnetic resonance imaging (MRI) with a diffusion-weighted imaging (DWI) sequence will confirm the presence of skin in the stenotic canal. Imaging findings include opacification (soft tissue filling) of the canal with bony remodeling of the surrounding bone.
As noted above, the inner ear develops much earlier in the fetus than the ear canal and middle ear. In addition, the inner ear develops from a completely separate structure from the middle and outer ears. As such, most patients with CAA have normal inner ear (cochlear) function. The problem is simply that the sound energy is not being transmitted or conducted to the inner ear. This is called a conductive hearing loss. Ways to improve the transmission of sound to the healthy inner ear include a bone-conducting hearing aid, an osseointegrated bone-conducting device, and surgery to open the ear canal and restore the natural sound-conducting mechanism to the inner ear (atresia repair or atresia surgery).
Epidemiology
Frequency
The incidence of CAA is 1 in 10,000-20,000 live births. Unilateral CAA is more common than bilateral cases with an approximate ratio of 3-4:1. For unknown reasons, right ears are more commonly affected than left ears, and boys more commonly than girls.
Etiology
Factors that cause the developmental sequence of the ear canal and middle ear to cease in CAA are not known. Regions adjacent to the ear canal, including the jaw bone (mandible) and course of the facial nerve (the nerve that moves the muscles of facial expression) may be affected as well. In general, the inner ear (cochlea, hearing nerve, balance system) is not affected because its development is from a completely separate set of structures from the ear canal and middle ear, and the development of the inner ear is complete by the time the ear canal begins to form. In general, the middle ear, ossicles, and the auricle are affected because their development is concurrent with that of the ear canal. Studies have linked mutations in chromosome 18 to some patients with aural atresia and normally developed auricles. [10, 11]
Pathophysiology
Intimate knowledge of the anatomy and development of the ear is essential to understanding the clinical manifestations of CAA. The development of the ear consists of a complex series of events. The inner ear, middle ear space, ossicles, ear canal, and auricle each are derived from the 3 embryonic germ cell layers (ie, ectoderm, mesoderm, endoderm). These structures assume individual paths of growth and maturation.
Embryology of the ear
The ear canal and middle ear develop from the branchial apparatus, a series of ectodermal outpouchings (arches) and indentations (clefts/grooves) on the lateral surface of the embryo that are first clearly seen at 24 days gestation. The formation of the ear canal begins with the invagination of the first branchial groove (ie, primary meatus). This area is located between the first branchial arch rostrally (toward the head) and the second branchial arch caudally (toward the tail).
The branchial groove invaginates and advances medially as an epithelial plate as early as the second month of fetal life. Its ingrowth temporarily meets the lateral growth of the first pharyngeal pouch. The first pharyngeal pouch is derived from endoderm of the foregut (alimentary canal) and subsequently develops into the middle ear cleft and Eustachian tube. The union between the ingrowth of the first branchial groove and first pharyngeal pouch forms the meatal plate, which is the precursor to the tympanic membrane. At 6 months of fetal life, the epithelial plate begins to canalize (open) from a medial to lateral direction (from the inside out) to meet the primary meatus.
At birth, the ear canal comprises the bony tympanic ring medially and a membranous cartilaginous portion laterally. After birth, the bony tympanic ring lengthens and transforms from a ring into a bony cylinder. Thus, the ear canal increases in length and reaches adult proportions by the age of 4-5 years.
In CAA, development of the ear may be interrupted at any point. If the process is halted before canalization of the ear canal, total atresia occurs. If development is halted during canalization, the patency of the external ear canal varies depending on how far canalization has progressed.
Development of the middle ear space and ear bones (ossicles) is closely aligned with the development of the ear canal, from the branchial apparatus. The middle ear space arises from lateral growth of the first pharyngeal pouch. The middle ear eventually envelops the ossicles and incorporates them into the middle ear space. The ossicles are derived from both first and second branchial arches, specifically Meckel and Reichert cartilages, respectively. Arrest in the development of the ear canal affects the middle ear to the extent that the growth and maturation of the middle ear will also be incomplete, and the middle ear space is constricted. The ossicular chain is usually deformed, typically featuring a fused malleus-incus complex, shortened malleus with absent manubrium, and occasionally, hypoplastic stapes suprastructure. The malleus neck may be fused to the bony atretic plate. In cases of complete atresia, the tympanic membrane is absent.
The inner ear begins development as early as the third week of fetal life with the formation of the otic placode, a local thickening of the ectoderm. The otic placode invaginates to form the otic pit. The epithelium of the otic pit fuses to become the otic vesicle, which forms the membranous labyrinth of the inner ear. A series of infoldings of the otic vesicle compartmentalizes the membranous labyrinth into the vestibule, cochlea, and endolymphatic regions. The inner ear completes its development by the 20th week of fetal life, which predates the formation of the ear canal and explains why patients with CAA generally have a cochlea that is functional and able to be stimulated.
CAA commonly coexists with microtia. However, rarely, CAA may occur alone with an auricle that appears healthy. The embryologic precursors of the auricle are the axonal hillocks, a series of ectodermal elevations derived from the first and second branchial arches located along the rostral end of the branchial apparatus. The axonal hillocks fuse with each other around the primitive ear canal. Each of the 6 hillocks is responsible for forming a distinctive part of the auricle. Hillock 1 forms the tragus; hillock 2 forms the crus helicis; hillock 3 forms the helix; hillock 4 forms the antihelix; hillock 5 forms the antitragus; and hillock 6 forms the ear lobule. The auricle assumes adult shape (but not size) by the 20th week of fetal life. Microtia ("small ear") results when the development of these hillocks is arrested. In general, the middle ear is less developed when microtia is severe than when it is not. [2]
Presentation
The patient with CAA may present with unilateral or bilateral atresia. If the deformity is present in both ears, the severity often varies between the ears. Hearing evaluation through auditory-evoked brainstem response (ABR; BAER) testing and hearing habilitation, generally with a bone-conducting hearing aid, are critical early in life for normal language development in children with bilateral CAA.
A study by Montino et al found that out of 68 children with CAA, 94.3% had age-adequate feeding abilities, 44.1% had language development delays, and, in patients with bilateral, right, or left CAA, 57.2%, 53.9%, and 53.4%, respectively, had orofacial development delays. [12]
Colman graded the severity of CAA into 3 categories: minor, moderate, and severe. Patients with severe CAA have total canal atresia and usually present with unfavorable middle ear anatomy and temporal bone development for reconstruction. In comparison, patients with moderate CAA have more favorable anatomy (ie, an identifiable ear canal with deformed ossicles and an aerated middle ear space). Most patients with CAA present with this moderate form. In minor cases, the ear canal is present but narrowed, and the middle ear is better developed.
CAA may coexist with syndromes that feature first and second branchial arch deformities (eg, Treacher-Collins syndrome, hemifacial microsomia, Goldenhar syndrome, and other craniofacial abnormalities).
The patient with CAA may present to the otologist in any of the scenarios discussed next.
Detection at birth
If the anatomic abnormality is detected at birth, the otologist examines the newborn and sees the parents in consultation. The parents are usually anxious and eager to have their many questions answered. Reassurance and parent education are critical during this time. Regardless of whether this condition is present in 1 or both ears, cochlear function must be assessed in both ears by means of specialized audiologic testing, such as auditory-evoked brainstem responses (ABR or BAER testing) elicited from both air-conducted and bone-conducted signals. If bilateral CAA and good cochlear function are present, the infant needs amplification with a bone-conducting hearing aid (eg, BAHA Softband) until a decision is made to pursue surgical correction around the age of 5-7.
The decision to amplify, or place a bone-conducting hearing device, on the child with unilateral CAA is one made individually by the family. There are advantages and disadvantages to using a bone conductor as well as to not using one. Physicians should never discourage families from trying a bone conductor on their child with unilateral CAA, but families should not be discouraged if their child refuses to wear it. While researchers are discovering some of the more subtle problems associated with unilateral hearing loss (which include difficulty hearing in noise and difficulty locating sound in space), especially in school-age children, [13] these studies have been conducted in children with unilateral sensorineural hearing loss, not conductive hearing loss. A subsequent report showed that none of the children with unilateral CAA who were studied repeated a grade in elementary education but that they did utilize resources such as an individual education program (IEP) and speech therapy to a greater degree. [14] In general, normal hearing in one ear is sufficient hearing to support normal speech and language development.
That said, it is critical in the early years to monitor the hearing in the normally developed ear to ensure that it is hearing well. Follow-up ABR testing is recommended every 3-4 months, and then every 6 months, in the first 1-2 years of life, followed by behavioral testing every 6 months in the second 2 years of life and, if the hearing is stable, annually thereafter.
Once in the elementary school years, the child should be monitored for academic success. Resources such as an IEP/504(c) plan, speech therapy, preferential seating in class, and FM systems should be instituted early to set the child up for success in the academic environment.
Detection in the older child just before microtia repair
The otologist may encounter the older child with unilateral CAA before a plastic surgeon repairs microtia. In these patients, coordination of care between the plastic surgeon and otologist is important to delineate a clear surgical plan and schedule to prevent confusion and unrealistic expectations. [3] Using the patient's own rib ("autologous"), the plastic surgeon sculpts an auricle and pockets it under the skin, moves the lobule, elevates the sculpted rib off the skull with a skin graft, and creates a tragus in a multistage (3-4 operations over a one year period) procedure. If the child is a candidate for atresia repair surgery, the otologist opens the new ear canal, frees the ossicular chain so the ear bones can vibrate, constructs a tympanic membrane, lines the ear canal with a skin graft, and opens the meatus in continuity with the new ear canal, in a single procedure.
A porous polyethylene implant (Medpor, Su-Por) has also been established as a viable means of reconstructing the auricle. Long-term (12 years) data show a complication rate of exposure of the implant at 7%. [15] The implant in the setting of atresia can be problematic if the atresia repair is performed after the implant has been placed. In this instance, the implant is at risk for exposure, infection, and extrusion. Patients have undergone atresia repair before Medpor implantation with short-term success. [16] Revision atresia repair in these patients again puts the Medpor implant at risk.
Clinical picture with canal stenosis
A child with canal stenosis (mild atresia) may present to the clinician with or without associated hearing loss in the ear. These patients with narrowed canals (generally less than 2 mm) are at risk for canal cholesteatoma. [17] Cholesteatoma is a pocket or cyst of skin that grows and expands as dead skin cells fill the cyst. Careful observation, follow-up, and microscopic examination and cleaning may ensure that the patient does not develop a canal cholesteatoma. However, when the opening to the ear canal is too small to permit examination or when the patient's history (eg, ear drainage or pain) suggests a cholesteatoma, radiographic imaging with CT scanning is used to evaluate the possibility of a canal cholesteatoma; CT scan image of a patient with canal cholesteatoma is seen below.
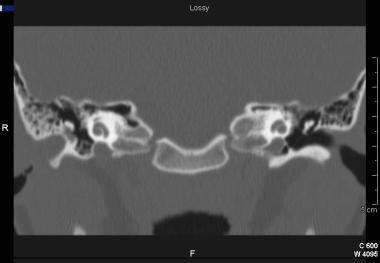 Coronal CT scan of patient with ear canal cholesteatoma (right ear) in the setting of congenital aural stenosis. Note the rounded edges of ear canal bone filled with soft tissue density extending into the middle ear.
Coronal CT scan of patient with ear canal cholesteatoma (right ear) in the setting of congenital aural stenosis. Note the rounded edges of ear canal bone filled with soft tissue density extending into the middle ear.
Children with canals 2 mm or smaller in diameter develop canal cholesteatoma more frequently than do other children. In addition, canal cholesteatomas are rarely, if ever, observed to occur in children younger than 3 years. Cole and Jahrsdoerfer reported these findings and noted that 50% of 54 stenotic ears with canals smaller than 4 mm developed canal cholesteatoma. [17] Clinicians should readily incorporate CT scanning into their diagnostic workup in children with stenotic ear canals to ensure that no canal cholesteatoma, medial to the stenosis, is present. CT scanning can be performed when the child is aged 4-5 years and should not be done during infancy. Canal cholesteatoma in the setting of complete aural atresia very rarely, if ever, occurs.
Clinical picture in the adult with CAA
Adults with CAA who present to the otologist usually have a unilateral condition that was not corrected in childhood. In some patients, the condition was unnoticed, especially in those with stenotic ears. If it was noticed, the patients may have been told that their condition was inoperable or too risky to repair in light of possible facial nerve injury. This is especially true if the contralateral ear was morphologically and functionally normal. In this situation, the patient may make an informed decision to proceed with an evaluation for surgery with dedicated CT scan and audiometry with the hopes of obtaining binaural hearing. However, these patients can also be offered bone conduction hearing technology as a viable alternative.
Indications
Any child with microtia/atresia and normal bone conduction thresholds (normal inner ear or cochlear function) is a candidate for habilitative resources. These resources may include preferential seating in school, an FM system in which the teacher wears a microphone and the child has a speaker at his or her desk or an ear bud, or a hearing aid. FM systems improve the signal-to-noise ratio for children with hearing loss and can be effective in the classroom setting.
Individualized education programs (IEPs, 504(c) plans) are also available for children with aural atresia. Speech therapy is also indicated if the expressive language is not progressing.
Amplification options include bone-conducting hearing systems such as the BAHA Softband (Cochlear; Sydney, Australia), a bone oscillator worn on a headband tight to the bone around the head to optimize bone conduction to the cochlea, or, when the child is aged 5 years or older, an osseointegrated bone-conduction hearing device, an implantable titanium post to which the bone oscillator is affixed. Osseointegrated bone-conducting systems in which a post (abutment) comes through the skin (percutaneous) include the BAHA Connect System (Cochlear; Sydney, Australia) and the Ponto System (Oticon Medical; Somerset, NJ). Osseointegrated bone-conducting systems in which a magnet attaches to the titanium implant completely under the skin (transcutaneous passive) include the BAHA Attract System (Cochlear) and the Sophono system (Sophono; Boulder, CO).
The latter two devices (Sophono and BAHA Attract) require surgical placement of a metallic plate into the bone of the skull behind the ear. Once healed, the sound processor is attached to a magnetic plate that is placed on the skin over the internal magnetic plate. Sound enters the processor, which vibrates, and through the magnetic field vibrates the underlying magnetic plate and implant placed in the bone, sending the sound energy to the cochlea via bone conduction.
The BAHA System (implanted titanium post) is not approved in the United States for children younger than 5 years. These devices utilize bone conduction, sending the sound energy through the bone of the skull directly to the cochlea and bypassing the outer and middle ear.
Finally, two newer devices (transcutaneous active) place the active vibrational stimulators under the skin. With the Cochlear Osia System (Cochlear) and the Bonebridge device (Med-El Medical Electronics; Innsbruck, Austria) the patient wears a microphone-transmitter on a magnet that sends the sound signal across the skin to the "actuator," or active piece secured to the bone. Both devices have shown favorable efficacy in hearing habilitation in patients with conductive hearing loss. [18, 19, 20]
Before surgical atresia repair (canal surgery) is considered, the patient must fulfill two criteria. First, the patient with CAA must have normal cochlear function, as demonstrated on audiologic examination with normal bone conduction thresholds in the atretic ear. Second, CT scanning must demonstrate normal inner ear and internal auditory canal morphology.
If patients meet these criteria, the next step is to identify patients with favorable anatomy who have a reasonable chance of success with surgical correction. Jahrsdoerfer devised a 10-point grading scheme, based on features from the CT scan and appearance of the external ear, that reflects the likelihood of success (see Imaging Studies). [21] Patients who have a score of 7 or higher are good candidates for surgery. Patients must also be motivated to undergo the surgery and postoperative care and must be willing to have the ear cleaned of dead skin once or twice a year for the rest of their lives.
Numerous studies now show that in patients with favorable anatomy, repair of unilateral CAA is a viable and successful undertaking. [22, 23]
Relevant Anatomy
Critical assessment of the anatomy is necessary in the evaluation and treatment of the patient with congenital aural atresia (CAA). Even if surgical correction is not sought, the otologist must understand the patient's anatomy and be able to assess the CT scan to explain the nature of the deformity to the parents or patient. Every CAA patient's anatomy is different.
The relevant anatomy includes the patency (or lack thereof) of the ear canal. In the absence of an ear canal, the atretic plate is the bony region just lateral to the middle ear space. Assessment of the middle ear space should include the appearance of each of the three ossicles and the incudostapedial joint (the joint between the second and third ear bones, the incus and stapes) and the position and course of the facial nerve through all of its segments. The course of the facial nerve is often aberrant in CAA, as the nerve typically courses more anterior and lateral in its proximal mastoid segment just after the second genu. In addition, the second turn takes a more acute bend than normal.
Patency of the round and oval windows must be ensured. The otologist should evaluate the size and pneumatization of the middle ear cavity and mastoid. Size, or volume, of the middle ear space has been shown to correlate with hearing outcomes; ie, the larger the middle ear volume, the better and more stable the hearing results. [24] Relationship to the jaw joint is also important to determine the amount of room for drilling the ear canal. The position of the tegmen (bone separating the ear from the brain) is important, as the "roof" of the ear may hang down too far inferiorly to open an ear canal. [25] Very low tegmen is seen in the image below. The morphology of the inner ear is also evaluated for concurrent inner ear dysplasia. [26]
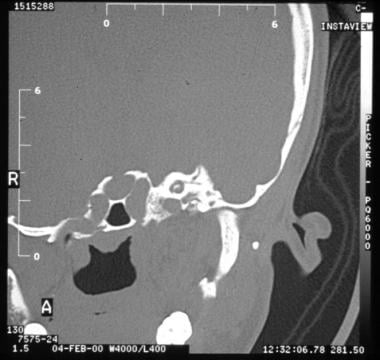 Very low tegmen (bone that separates brain from ear) and lack of middle ear aeration make this patient not a candidate for atresia surgery.
Very low tegmen (bone that separates brain from ear) and lack of middle ear aeration make this patient not a candidate for atresia surgery.
Contraindications
An absolute contraindication to surgical correction of congenital aural atresia (CAA) is lack of cochlear function in the involved ear as documented on audiological examination. In this condition, bone conduction thresholds will be in the moderate-severe-profound range. CT scan may reveal a dysplastic or aplastic inner ear (rare). Surgical correction of CAA is pointless if the patient has no cochlear function. However, if microtia is present, reconstruction of the auricle may be undertaken for cosmetic purposes.
Lack of middle ear aeration is another contraindication to surgery. Without an air space, the ossicles cannot vibrate, and the sound conducting mechanism of the middle ear will not be restored.
The tegmen is the bone that separates the brain from the ear. If the tegmen hangs too low, not enough space exists to open an ear canal; brain injury is a risk during the drilling. The patient is not a candidate for atresia surgery (see the image below).
Very low tegmen (bone that separates brain from ear) and lack of middle ear aeration make this patient not a candidate for atresia surgery.
Although some authors have advocated transposition of the facial nerve in some situations, a facial nerve coursing over the oval window or in direct line of canal drilling would be a relative contraindication. [27]
-
Grade III microtia with complete absence (atresia) of the external auditory canal.
-
Appearance of surgically-corrected microtia with complete atresia of the external ear canal.
-
Axial CT scan in a patient with congenital aural atresia (CAA). Note the atretic plate just lateral to the ossicles.
-
Coronal CT scan of left temporal bone in a patient with congenital aural atresia (CAA). Note absence of the ear canal. The atretic plate may be seen just lateral to the ossicles.
-
Three-dimensional reconstructed CT scan of lateral skull in a child with aural atresia. Note the absence of a bony aperture (external ear canal) on the side of the left temporal bone.
-
Coronal CT scan of patient with ear canal cholesteatoma (right ear) in the setting of congenital aural stenosis. Note the rounded edges of ear canal bone filled with soft tissue density extending into the middle ear.
-
Very low tegmen (bone that separates brain from ear) and lack of middle ear aeration make this patient not a candidate for atresia surgery.
-
Intraoperative photograph of a newly drilled ear canal, middle ear space, and ossicular chain in a patient with congenital aural atresia (CAA).
-
Intraoperative image of the bony landmarks used to identify the site for drilling the bony canal in a right ear - zygomatic root, glenoid fossa, mastoid tip. Patient in the surgical position.
-
Intraoperative image of a left ear in the surgical position upon entering the middle ear in the epitympanum. The fused malleus-incus complex can be seen.
-
Intraoperative image of a right ear with a lateral facial nerve identified in the mastoid segment.
-
Intraoperative image of the right ear with the fascia graft draped over the ossicular chain
-
Intraoperative image of a right ear with the skin graft placed in position
-
Intraoperative image of a right ear meatoplasty.
-
Axial CT scan of a right ear canal cholesteatoma. Note opacification along a bony track up to the ossicles.





