Overview
Healing...is not a science but the intuitive art of wooing nature.
W.H. Auden, "The Art of Healing"
Wound healing is a natural restorative response to tissue injury. Healing is the interaction of a complex cascade of cellular events that generates resurfacing, reconstitution, and restoration of the tensile strength of injured skin.
Healing is a systematic process, traditionally explained in terms of 4 overlapping classic phases: hemostasis, inflammation, proliferation, and maturation. While platelets play a crucial role in clot formation during hemostasis, inflammatory cells débride injured tissue during the inflammatory phase. Epithelialization, fibroplasia, and angiogenesis occur during the proliferative phase. Meanwhile, granulation tissue forms and the wound begins to contract. Finally, during the maturation phase, collagen forms tight cross-links to other collagen and with protein molecules, increasing the tensile strength of the scar. [1]
For the sake of discussion and understanding, the process of wound healing may be presented as a series of separate events. In actuality, the entire process is much more complicated, as cellular events that lead to scar formation overlap. Many aspects of wound healing have yet to be elucidated.
An image depicting a hypertrophic scar can be seen below.
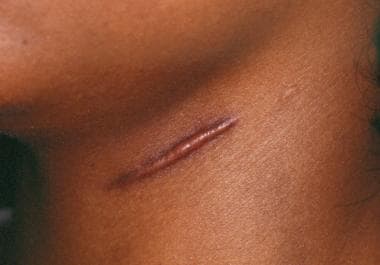 Hypertrophic scarring and keloids. Hypertrophic scars on neck with raised features within scar margins.
Hypertrophic scarring and keloids. Hypertrophic scars on neck with raised features within scar margins.
Hemostasis
Hemostasis starts within the very first moments of the injury, where platelets play a role by aggregating, as well as by releasing cytokines, chemokines, and hormones.
Vasoconstriction occurs to limit blood loss under the effects of vasoactive mediators (epinephrine, norepinephrine, prostaglandins, serotonin, and thromboxane), causing temporary blanching of the wound.
Exposed subendothelium, including collagen and tissue factor, activates platelet aggregation and leads to clot formation (primary plug).
Aggregation and attachment to exposed collagen surfaces activates the platelets. Activation enables platelets to degranulate and release chemotactic and growth factors, such as platelet-derived growth factor (PDGF), proteases, and vasoactive agents (eg, serotonin, histamine).
Chemokines released by platelet activation attract inflammatory cells to the area, leading to the next phase in the healing process.
Inflammatory Phase
In addition to activation of fibrin, thrombin activated by the coagulation cascades facilitates migration of inflammatory cells to the site of injury by increasing vascular permeability. By this mechanism, factors and cells necessary for healing flow from the intravascular space and into the extravascular space.
At this time, clot formation dissipates as its stimuli dissipate. Plasminogen is converted to plasmin, a potent enzyme that aids in cell lysis.
The initial vasoconstriction period, typically only 10-15 minutes in duration, is followed by a more persistent period of vasodilation mediated by histamine, prostaglandins, kinins, and leukotrienes. Vasodilation is an important means by which the wound can be exposed to increased blood flow, accompanied by the necessary inflammatory cells and factors that fight infection and débride the wound of devitalized tissue. Alterations in pH (secondary to tissue and bacterial degradation), swelling, and tissue hypoxemia at the injury site contribute to the sensation of wound pain.
The cellular aspect of the inflammatory phase occurs within hours of injury, and it includes neutrophils, macrophages, and lymphocytes.
Neutrophils are the predominant cell type for the first 48 hours after injury but do not appear essential to the wound-healing process. Neutrophils cleanse the wound site of bacteria and necrotic matter and release inflammatory mediators and bactericidal oxygen free radicals. The absence of neutrophils does not prevent healing.
Macrophages are essential to wound healing and perhaps are the most important cells in the early phase of wound healing. [2] Macrophages phagocytose debris and bacteria. Macrophages also secrete collagenases and elastases, which break down injured tissue and release cytokines. In addition, macrophages release PDGF, an important cytokine that stimulates the chemotaxis and proliferation of fibroblasts and smooth muscle cells. Finally, macrophages secrete substances that attract endothelial cells to the wound and stimulate their proliferation to promote angiogenesis. Macrophage-derived growth factors play a pivotal role in new tissue formation, as evidenced by the fact that new tissue formation in macrophage-depleted animal wounds demonstrates defective repair. In studies in which experimental wounds are rendered monocytopenic, subsequent stages of fibroplasia and granulation tissue formation are impaired and the overall rate of wound healing is delayed.
T lymphocytes migrate into the wound during the inflammatory phase, approximately 72 hours following injury. T lymphocytes are attracted to the wound by the cellular release of interleukin 1, which also contributes to the regulation of collagenase. Lymphocytes secrete lymphokines such as heparin-binding epidermal growth factor and basic fibroblast growth factor. Lymphocytes also play a role in cellular immunity and antibody production.
Proliferative Phase
Formation of granulation tissue is a central event during the proliferative phase. Its formation occurs 3-5 days following injury and overlaps with the preceding inflammatory phase. Granulation tissue includes inflammatory cells, fibroblasts, and neovasculature in a matrix of fibronectin, collagen, glycosaminoglycans, and proteoglycans.
Epithelialization
Epithelialization is the formation of epithelium over a denuded surface. It involves the migration of cells at the wound edges over a distance of less than 1 mm, from one side of the incision to the other. Incisional wounds are epithelialized within 24-48 hours after injury. This epithelial layer provides a seal between the underlying wound and the environment.
The process begins within hours of tissue injury. Epidermal cells at the wound edges undergo structural changes, allowing them to detach from their connections to other epidermal cells and to their basement membrane. Intracellular actin microfilaments are formed, allowing the epidermal cells to creep across the wound surface. As the cells migrate, they dissect the wound and separate the overlying eschar from the underlying viable tissue. In superficial wounds (eg, wounds due to laser resurfacing, dermabrasion, chemical peel treatments) adnexal structures (eg, sebaceous glands, hair follicles) contribute to reepithelialization.
Epidermal cells secrete collagenases that break down collagen and plasminogen activator, which stimulates the production of plasmin. Plasmin promotes clot dissolution along the path of epithelial cell migration. Migrating epithelial cells interact with a provisional matrix of fibrin cross-linked to fibronectin and collagen. In particular, fibronectin seems to promote keratinocyte adhesion to guide these cells across the wound base.
Wounds in a moist environment demonstrate a faster and more direct course of epithelialization. Occlusive and semiocclusive dressings applied in the first 48 hours after injury may maintain tissue humidity and optimize epithelialization. Studies have shown that occlusive dressings in the form of silicone sheeting, or other forms of occlusive dressing, limit epidermal inflammatory factors that contribute to epidermal hyperplasia, by restoring barrier function and increasing skin hydration. [3]
When epithelialization is complete, the epidermal cell assumes its original form, and new desmosomal linkages to other epidermal cells and hemidesmosomal linkages to the basement membrane are restored.
Fibroplasia
The fibroblast is a critical component of granulation tissue. Fibroblasts are responsible for the production of collagen, elastin, fibronectin, glycosaminoglycans, and proteases. Fibroblasts grow in the wound as the number of inflammation cells decrease. The demand for inflammation disappears as the chemotactic factors that call inflammatory cells to the wound are no longer produced and as those already present in the wound are inactivated.
Fibroplasia begins 3-5 days after injury and may last as long as 14 days. Skin fibroblasts and mesenchymal cells differentiate to perform migratory and contractile capabilities. Fibroblasts migrate and proliferate in response to fibronectin, platelet-derived growth factor (PDGF), fibroblast growth factor, transforming growth factor, and C5a. Fibronectin serves as an anchor for the myofibroblast as it migrates within the wound.
The synthesis and deposition of collagen is a critical event in the proliferative phase and to wound healing in general. Collagen is rich in hydroxylysine and hydroxyproline moieties, which enable it to form strong cross-links. The hydroxylation of proline and lysine residues depends on the presence of oxygen, vitamin C, ferrous iron, and α-ketoglutarate. Deficiencies of oxygen and vitamin C, in particular, result in underhydroxylated collagen that is less capable of forming strong cross-links and, therefore, is more vulnerable to breakdown.
Collagen is secreted to the extracellular space in the form of procollagen. This form is then cleaved of its terminal segments and called tropocollagen. Tropocollagen can aggregate with other tropocollagen molecules to form collagen filaments.
Filament, fibril, and fiber formation occur within a matrix gel of glycosaminoglycans, hyaluronic acid, chondroitin sulfate, dermatan sulfate, and heparin sulfate produced by fibroblasts.
Intermolecular cross-links within the collagen fiber stabilize it, making it resistant to destruction. Age, tension, pressure, and stress affect the rate of collagen synthesis. Collagen synthesis begins approximately 3 days after injury and may continue at a rapid rate for approximately 2-4 weeks. Collagen synthesis is controlled by the presence of collagenases and other factors that destroy collagen as new collagen is made.
Approximately 80% of the collagen in normal skin is type I collagen; the remaining is mostly type III. In contrast, type III collagen is the primary component of early granulation tissue and is abundant in embryonic tissue. Collagen fibers are deposited in a framework of fibronectin. An essential interaction seems to exist between fibronectin and collagen; experimental wounds depleted of fibronectin demonstrate decreased collagen accumulation.
Elastin is also present in the wound in smaller amounts. Elastin is a structural protein with random coils that allow for stretch and recoil properties of the skin.
Angiogenesis
A rich blood supply is vital to sustain newly formed tissue and is appreciated in the erythema of a newly formed scar. The macrophage is essential to the stimulation of angiogenesis and produces macrophage-derived angiogenic factor in response to low tissue oxygenation. This factor functions as a chemoattractant for endothelial cells. Basic fibroblast growth factor secreted by the macrophage and vascular endothelial growth factor secreted by the epidermal cell are also important to angiogenesis.
Angiogenesis results in greater blood flow to the wound and, consequently, increased perfusion of healing factors. Angiogenesis ceases as the demand for new blood vessels ceases. New blood vessels that become unnecessary disappear by apoptosis.
Research indicates that at each phase of the wound healing process, host defense peptides (HDPs) are essential. For example, the HDP LL-37, a cathelicidin, has been found to aid dermal neovascularization during the proliferative phase, stimulating endothelial cells and thus driving up the proliferation rate. [4]
Contraction
Wound contraction, defined as the centripetal movement of wound edges that facilitates closure of a wound defect, essentially begins concurrent with collagen synthesis, and reaches maximal activity at 5-15 days after injury. Contraction results in a decrease in wound size, appreciated from end to end along an incision; a 2-cm incision may measure 1.8 cm after contraction. The maximal rate of contraction is 0.75 mm/d and depends on the degree of tissue laxity and shape of the wound. Loose tissues contract more than tissues with poor laxity, and square wounds tend to contract more than circular wounds. Wound contraction depends on the myofibroblast located at the periphery of the wound, its connection to components of the extracellular matrix, and myofibroblast proliferation.
Radiation and drugs, which inhibit cell division, have been noted to delay wound contraction. Contraction does not seem to depend on collagen synthesis.
Although the role of the peripheral nervous system in wound healing is not well delineated, recent studies have suggested that sympathetic innervation may affect wound contraction and epithelialization through unknown mechanisms.
Contraction must be distinguished from contracture, a pathologic process of excessive contraction that limits motion of the underlying tissues and is typically caused by the application of excessive stress to the wound.
Maturation Phase
Collagen
Collagen remodeling during the maturation phase depends on continued collagen synthesis in the presence of collagen destruction. Collagenases and matrix metalloproteinases in the wound assist removal of excess collagen while synthesis of new collagen persists. Tissue inhibitors of metalloproteinases limit these collagenolytic enzymes, so that a balance exists between formation of new collagen and removal of old collagen.
During remodeling, collagen becomes increasingly organized. Fibronectin gradually disappears, and hyaluronic acid and glycosaminoglycans are replaced by proteoglycans. Type III collagen is replaced by type I collagen. Water is resorbed from the scar. These events allow collagen fibers to lie closer together, facilitating collagen cross-linking and ultimately decreasing scar thickness. Intramolecular and intermolecular collagen cross-links result in increased wound bursting strength. Remodeling begins approximately 21 days after injury, when the net collagen content of the wound is stable. Remodeling may continue indefinitely.
The tensile strength of a wound is a measurement of its load capacity per unit area. The bursting strength of a wound is the force required to break a wound regardless of its dimension. Bursting strength varies with skin thickness. Peak tensile strength of a wound occurs approximately 60 days after injury. A healed wound only reaches approximately 80% of the tensile strength of unwounded skin.
Cytokines
Cytokines have emerged as important mediators of wound healing events. By definition, a cytokine is a protein mediator, released from various cell sources, which binds to cell surface receptors to stimulate a cell response. Cytokines can reach their target cell by endocrine, paracrine, autocrine, or intracrine routes. Some important cytokines are described as follows:
-
Epidermal growth factor was the first cytokine described and is a potent mitogen for epithelial cells, endothelial cells, and fibroblasts. Epidermal growth factor stimulates fibronectin synthesis, angiogenesis, fibroplasia, and collagenase activity.
-
Fibroblast growth factor is a mitogen for mesenchymal cells and an important stimulus for angiogenesis. Fibroblast growth factor is a mitogen for endothelial cells, fibroblasts, keratinocytes, and myoblasts. This factor also stimulates wound contraction and epithelialization and production of collagen, fibronectin, and proteoglycans.
-
PDGF is released from the alpha granules of platelets and is responsible for the stimulation of neutrophils and macrophages and for the production of transforming growth factor-β. PDGF is a mitogen and chemotactic agent for fibroblasts and smooth muscle cells and stimulates angiogenesis, collagen synthesis, and collagenase. Vascular endothelial growth factor is similar to PDGF but does not bind the same receptors. Vascular endothelial growth factor is mitogenic for endothelial cells and plays an important role in angiogenesis.
-
Transforming growth factor-β is released from the alpha granules of platelets and has been shown to regulate its own production in an autocrine manner. This factor is an important stimulant for fibroblast proliferation and the production of proteoglycans, collagen, and fibrin. The factor also promotes accumulation of the extracellular matrix and fibrosis. Transforming growth factor-β has been demonstrated to reduce scarring and to reverse the inhibition of wound healing by glucocorticoids.
-
Tumor necrosis factor-α is produced by macrophages and stimulates angiogenesis and the synthesis of collagen and collagenase. Tumor necrosis factor-α is a mitogen for fibroblasts.
A literature review by Russo et al reported strong evidence that keratinocyte-produced interleukin 1 (IL-1) plays a part in inducing fibroblasts to produce fibroblast growth factor 7 (FGF-7; also called keratinocyte growth factor [KGF]), granulocyte-monocyte colony stimulating factor (GM-CSF), transforming growth factor-α (TGF-α), IL-6, IL-8, and IL-1, as well as the expression of cyclooxygenase 2 (COX2) and prostaglandin E2 (PGE2) production. FGF-7, GM-CSF, and PGE2, in turn, promote the proliferation of keratinocytes, with proper keratinocyte differentiation favored. [5]
Primary and Secondary Healing
By definition, primary healing involves uncomplicated healing of noninfected, well-approximated wounds. The 4 phases are respected in this process without interruption. [6]
Healing by secondary intention involves generation of granulation tissue and epithelialization over it. Given the fact that angiogenesis and epithelialization take longer in secondary intention healing, these wounds are more prone to infection and poor healing.
When properly monitored, healing by secondary intention may lead to acceptable cosmetic results in some areas of the body. When considering secondary intention for wound healing, certain factors need to be considered, including surrounding skin pigmentation and location on the face. The pigmentation of the surrounding skin is important because melanocytes do not migrate into healing wounds; thus, areas left to granulate will be hypopigmented relative to the surrounding unaffected tissue. The best locations for healing by secondary intention include areas with lax skin and concavities, such as the concave surfaces of the eye, ear, and nose. [7, 8]
Abnormal Wound Healing: Keloids and Hypertrophic Scars
Keloids and hypertrophic scars are characterized by an accumulation of excess collagen with decreased lysis and are distinguished from each other by their appearance. Keloids grow beyond the borders of the original wound and do not tend to resolve spontaneously. Hypertrophic scars stay within the limit of the original wound and do tend to regress spontaneously. Hypertrophic scars are generally seen soon after tissue injury, whereas keloids can form as late as a year after injury. Hypertrophic scars tend to be associated with a contracture across a joint surface.
Upon histological examination, keloids and hypertrophic scars differ from normal skin by a rich blood supply, high mesenchymal density, and a thick epidermal layer. The collagen fibers in hypertrophic scars are loosely arrayed in a wavy pattern. Keloids demonstrate a disorganized pattern of large, irregularly shaped collagen fibers with a lower content of collagen cross-links compared to normal skin. Keloids also contain a greater amount of type III collagen than a mature scar, which suggests a failure in scar maturation.
Little is known about the etiology of keloid and hypertrophic scar formation. Abnormalities in cell migration, proliferation, inflammation, and the synthesis and secretion of extracellular matrix proteins, cytokines, and remodeling all have been associated with keloid and hypertrophic scar formation. Increased activity of fibrogenic cytokines and exaggerated responses to cytokines have been described. Transforming growth factor-β (TGF-β) appears to play an integral role in aberrant healing. More recently, abnormal interactions between epidermal-mesenchymal cells and regulatory genes, such as TP53, have been proposed.
Research by Yamawaki et al indicates that the serine protease HtrA1 may be tied to keloid development, with the investigators suggesting that the enzyme accelerates cell proliferation and is involved in the remodeling of keloid-specific extracellular matrix or cell surface molecules. [9]
Higher risk of hypertrophic scars and keloids exists in dark-skinned individuals, wounds that cross skin tension lines, and wounds that are located on the ear lobes or presternal and deltoid areas. A literature review by Butzelaar et al indicated that young age and bacterial colonization are also risk factors for hypertrophic scar formation, while chemotherapy, statin use, and smoking may be protective against such scarring. [10]
Of note, keloids have not yet been reported in persons with albinism. The role of wound tension has been examined and found to be related to the exaggerated response to tension of keloid fibroblasts relative to normal fibroblasts with regard to production of profibrotic growth factors.
A single, optimal treatment technique for hypertrophic scars and keloids has not been developed, and the recurrence rate of these abnormal scars is high. Conservative management includes pharmacologic therapy, pressure, laser, and radiotherapy. [11] Each method has varying degrees of reported success. Surgical management is reserved for cases that are unresponsive to conservative management.
Diabetes and Wound Healing
Diabetes has multiple effects on wound healing.
Microangiopathic disease results in impaired tissue blood flow and subsequent poor tissue oxygen delivery. Peripheral neuropathy predisposes to tissue trauma and an increased risk for infection. In addition, decreased immune function, slower collagen synthesis and accumulation, decreased angiogenesis, and poorer tensile strength of wounds leads to a high risk of wound dehiscence. [12]
Adequate control of blood glucose levels plays a crucial role in diabetic healing wound. Although definite parameters have not been defined, glucose levels of greater than 200 mg/dL are associated with worse outcomes. [13]
Effects of Smoking on Wound Healing
Smoking has a known negative effect on healing due to a direct toxic effect and the vasoconstriction induced by nicotine.
In 2003, Sorenson et al [14] followed 78 smokers for 15 weeks. They showed that the infection rate in smokers was 12%, compared with 2% in nonsmokers. They also stated that abstinence for 4 weeks prior to wounding resulted in a dramatic decrease in infection but no change in wound dehiscence. Interestingly, nicotine patch users had no adverse events.
Smoking has also been shown to decrease the function of neutrophils, inhibit collagen synthesis, and increase levels of carboxy-hemoglobin, owing to the effects of carbon monoxide and hydrogen cyanide.
A prospective cohort study by Lassig et al indicated that smoking negatively impacts wound healing’s inflammatory phase. The investigators found that in current/former smokers, postsurgical drain fluid from head and neck surgical wounds contained significantly lower concentrations of epidermal growth factor and soluble feline McDonough sarcoma (FMS)–like tyrosine kinase-1 than did wound fluid from patients who had never smoked. The study also found evidence that smoking impedes cutaneous perfusion in wound healing. [15]
In current practice, most surgeons insist on abstinence for at least 4-6 weeks before and after cosmetic surgery. Abstinence from smoking with nicotine replacement for 4 weeks prior to surgery has shown normalization of metalloproteinase levels to those of nonsmokers and higher levels of type I collagen. [16, 17, 18] A literature review by Pluvy et al found that smokers undergoing cosmetic surgery had a combined odds ratio of 2.3 for surgical site infections and 2.5 for delayed wound healing; for smokers undergoing bariatric surgery, combined odds ratios were 3.3 for delayed wound healing and 3.1 for cutaneous necrosis. [19]
Other factors contributing to poor wound healing include nutritional deficiency, prior radiation therapy, active wound infection, and tissue hypoxia. [20]
Future Research and Fetal Wound Healing
Wounds occurring in fetuses of early gestational age can heal without any scar formation. The difference in the wound environment in fetal wounds occurring in early gestation has been speculated to account for the absence of scar formation. Proposed contributing factors to scarless healing in fetal wounds are the presence of fewer neutrophils and more monocytes during the inflammatory period, different concentrations of cytokines, and a greater proportion of type III collagen in contrast to adult wounds. Although evidence indicates that early fetal wound models heal more efficiently than adult wounds, the explanation of this more-efficient process is under investigation. The explanation may also lie in alterations of the wound environment to mimic fetal wound models.
Evidence shows that the healing environment may be manipulated in other ways to enhance or to accelerate the healing process. Animal studies have demonstrated beneficial effects of both laser and electrical stimulation of tissues during the inflammatory, proliferative, and maturation phases of wound healing. Based on the assumption that proteins found in the blood can promote healing, the application of platelet-rich plasma has been investigated and found to foster increased levels of inflammatory cells during the healing process without affecting final cell counts in healed tissue.
A study by Misiura et al suggests that platelet-rich plasma promotes wound healing by activating a complex of growth factors and adhesion receptors, so that cell proliferation, migration, and collagen biosynthesis are stimulated. According to the study’s findings, activation of the epidermal growth factor receptor via platelet-rich plasma brings about peptidase-D–dependent proliferation of human keratinocytes. [21]
In healthy individuals, platelet-rich plasma may accelerate wound closure in full-thickness dermal wounds. However, it does not alter the long-term architecture of a healed wound. [22] Platelet-derived growth factor D alone may increase the rate of macrophage recruitment and angiogenesis, but its overall long-term effect is not established. [23]
A literature review by Seth et al looking at the effect of nutrition on surgical wound healing reported that enhanced wound healing and immune function are associated with intake of omega-3 fatty acids and specific amino acids, including glutamine. Moreover, wound-healing stages were found to be positively impacted by vitamins A, B, and C and by zinc, while results concerning the effects of vitamin E were variable. It was further indicated that polyphenolic compounds can aid recovery through their anti-inflammatory function. The investigators also stated that increased postoperative complications and infections could be linked to malnutrition and that “preoperative nutritional support correlated with reduced hospital stays and complications.” [24]
-
Hypertrophic scarring and keloids. Hypertrophic scars on neck with raised features within scar margins.








