Practice Essentials
Ventricular tachycardia (VT) or ventricular fibrillation (VF) is responsible for most of the sudden cardiac deaths in the United States, [1] at an estimated rate of approximately 300,000 deaths per year. [2, 3] VT refers to any rhythm faster than 100 (or 120) beats/min, with three or more irregular beats in a row, arising distal to the bundle of His. The rhythm may arise from the working ventricular myocardium, the distal conduction system, or both. See the image below.
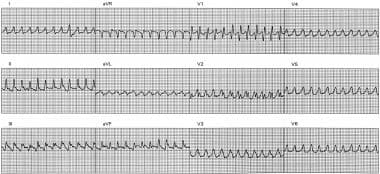 This electrocardiogram (ECG) shows rapid monomorphic ventricular tachycardia (VT), 280 beats/min, associated with hemodynamic collapse. The tracing was obtained from a patient with severe ischemic cardiomyopathy during an electrophysiologic study. A single external shock subsequently converted VT to sinus rhythm. The patient had an atrial rate of 72 beats/min (measured with intracardiac electrodes; not shown). Although ventriculoatrial dissociation (faster V rate than A rate) is diagnostic of VT, surface ECG findings (dissociated P waves, fusion or capture beats) are present in only about 20% of cases. In this tracing, the ventricular rate is simply too fast for P waves to be observed. VT at 240-300 beats/min is often termed ventricular flutter.
This electrocardiogram (ECG) shows rapid monomorphic ventricular tachycardia (VT), 280 beats/min, associated with hemodynamic collapse. The tracing was obtained from a patient with severe ischemic cardiomyopathy during an electrophysiologic study. A single external shock subsequently converted VT to sinus rhythm. The patient had an atrial rate of 72 beats/min (measured with intracardiac electrodes; not shown). Although ventriculoatrial dissociation (faster V rate than A rate) is diagnostic of VT, surface ECG findings (dissociated P waves, fusion or capture beats) are present in only about 20% of cases. In this tracing, the ventricular rate is simply too fast for P waves to be observed. VT at 240-300 beats/min is often termed ventricular flutter.
Signs and symptoms
History
Symptoms of VT are often a function of the associated heart rate, or the causal process, such as an acute myocardial infarction (MI). They may include the following bulleted items. VT may also be asymptomatic, or the symptoms may be those of the associated triggered therapy (eg, an implantable cardioverter-defibrillator [ICD] shock).
-
Palpitations
-
Light-headedness
-
Syncope
-
Chest pain
-
Anxiety
Physical examination
During VT, the following may be observed:
-
Hypotension
-
Tachypnea
-
Signs of diminished perfusion, including a diminished level of consciousness, pallor, and diaphoresis
-
High jugular venous pressure
-
Cannon A waves (if the atria are in sinus rhythm)
-
Variation in intensity of first heart sound (S1), caused by loss of atrioventricular (AV) synchrony
After cardioversion, physical findings during normal sinus rhythm are related to any underlying structural heart disease.
VT can also result in sudden death, especially after degeneration to VF. Patients in whom this occurs may first present with syncope.
See Presentation for more detail.
Diagnosis
Electrocardiography (ECG) is the criterion standard for the diagnosis of VT. If the clinical situation permits, a 12-lead ECG should be obtained before conversion of the rhythm. In a patient who is hemodynamically unstable or unconscious, however, the diagnosis of VT is made from the physical findings and ECG rhythm strip only. Advanced cardiovascular life support (ACLS) protocols should be quickly followed. Typically, laboratory tests should be deferred until electrical cardioversion has restored sinus rhythm and the patient is stabilized.
Assess levels of serum electrolytes, including the following, in all patients with VT:
-
Calcium (ionized calcium levels are preferred to total serum calcium levels)
-
Magnesium
-
Phosphate
Hypokalemia, hypomagnesemia, and hypocalcemia may predispose patients to either monomorphic VT or torsade de pointes.
Laboratory studies can also include the following:
-
Levels of therapeutic drugs (eg, digoxin)
-
Toxicology screens (potentially helpful in cases related to recreational or therapeutic drug use, such as cocaine or methadone)
-
Serum cardiac troponin I or T levels or other cardiac markers (to evaluate for myocardial ischemia or MI)
Postconversion VT
In patients with VT after conversion, the diagnostic workup proceeds as follows:
-
Repeat the ECG after termination of VT
-
Include electrolyte levels in an acute evaluation; the hyperadrenergic state or hemodynamic compromise often associated with VT may affect the subsequently obtained laboratory values
-
Perform toxicology screens for cocaine metabolites and tricyclic antidepressants, in accordance with the patient’s clinical history
-
Check cardiac enzyme levels if clinical symptoms or signs of ischemia are present
-
Perform echocardiography and coronary angiography after conversion to sinus rhythm to assess for structural and ischemic heart disease
Electrophysiologic study
Diagnostic electrophysiologic study (EPS) requires placement of electrode catheters in the ventricle, followed by programmed ventricular stimulation using progressive pacing protocols. EPS is particularly relevant in patients considered to be at high risk for sudden death as a result of significant underlying structural heart disease.
See Workup for more detail.
Management
Unstable patients with monomorphic VT should be immediately treated with synchronized direct current (DC) cardioversion, usually at a starting energy dose of 100 J (monophasic). Unstable polymorphic VT is treated with immediate defibrillation. Please refer to the most current ACLS guidelines, which are subject to periodic revision.
Medications
-
In stable patients with monomorphic VT and normal left ventricular function, restoration of sinus rhythm is typically achieved with intravenous (IV) procainamide, amiodarone, sotalol, or lidocaine
-
IV lidocaine is effective at suppressing peri-infarction VT but may have common and limiting side effects and, consequently, increase the overall mortality risk
-
In torsade de pointes, magnesium sulfate may be effective if a long QT interval is present at baseline
-
For long-term treatment of most patients with left ventricular dysfunction, current clinical practice favors class III antiarrhythmics (eg, amiodarone, sotalol)
-
In patients with heart failure, the best proven antiarrhythmic drug strategies include the use of beta receptor–blocking drugs (eg, carvedilol, metoprolol, bisoprolol); angiotensin-converting enzyme (ACE) inhibitors; and aldosterone antagonists
Implantable cardioverter-defibrillators
Multisociety guidelines recommend ICD therapy to augment medical management for the following [4] :
-
Most patients with hemodynamically unstable VT
-
Most patients with prior MI and hemodynamically stable sustained VT
-
Most cardiomyopathy patients with unexplained syncope (an arrhythmia is presumed)
-
Most patients with genetic sudden death syndromes when unexplained syncope is noted
Ablation
Radiofrequency ablation (RFA) via endocardial or epicardial catheter placement can be used to treat VT in patients who have the conditions noted in the following bulleted list. For patients with structural heart disease, it is currently uncertain whether VT ablation obviates other therapies, such as an ICD. [5, 6, 7, 8]
-
Left ventricular dysfunction from prior MI
-
Cardiomyopathy
-
Bundle-branch reentry
-
Various forms of idiopathic VT
See Treatment and Medication for more detail.
Background
Ventricular tachycardia (VT) refers to any rhythm faster than 100 (or 120) beats/min arising distal to the bundle of His. It is the most common form of wide complex tachycardia, with a high associated mortality rate. [9] The rhythm may arise from the working ventricular myocardium, the distal conduction system, or both. (See Etiology.) VT can be classified as sustained or nonsustained, with a generally accepted cutoff of 30 seconds.
VT is further classified according to the electrocardiographic (ECG) appearance. If the QRS complex remains identical from beat to beat, as occurs when VT originates from a single focus or circuit, it is classified as monomorphic (see the first two images below). If the QRS morphology changes from beat to beat, the VT is classified as polymorphic (see the third image below). Further classification can be made on the basis of the substrate and the location of the earliest activation.
This electrocardiogram (ECG) shows rapid monomorphic ventricular tachycardia (VT), 280 beats/min, associated with hemodynamic collapse. The tracing was obtained from a patient with severe ischemic cardiomyopathy during an electrophysiologic study. A single external shock subsequently converted VT to sinus rhythm. The patient had an atrial rate of 72 beats/min (measured with intracardiac electrodes; not shown). Although ventriculoatrial dissociation (faster V rate than A rate) is diagnostic of VT, surface ECG findings (dissociated P waves, fusion or capture beats) are present in only about 20% of cases. In this tracing, the ventricular rate is simply too fast for P waves to be observed. VT at 240-300 beats/min is often termed ventricular flutter.
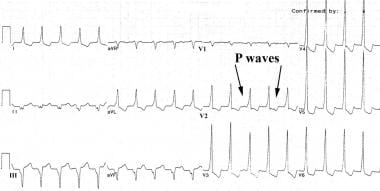 This electrocardiogram shows slow monomorphic ventricular tachycardia (VT), 121 beats/min, from a patient with an old inferior wall myocardial infarction and well-preserved left ventricular (LV) function (ejection fraction, 55%). The patient presented with symptoms of palpitation and neck fullness. Note the ventriculoatrial dissociation, which is most obvious in leads V2 and V3. Slower VT rates and preserved LV function are associated with better long-term prognosis.
This electrocardiogram shows slow monomorphic ventricular tachycardia (VT), 121 beats/min, from a patient with an old inferior wall myocardial infarction and well-preserved left ventricular (LV) function (ejection fraction, 55%). The patient presented with symptoms of palpitation and neck fullness. Note the ventriculoatrial dissociation, which is most obvious in leads V2 and V3. Slower VT rates and preserved LV function are associated with better long-term prognosis.
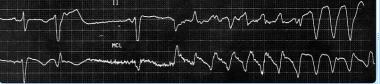
In the United States, the most common setting for VT is ischemic heart disease, in which myocardial scar tissue is the substrate for electrical reentry. VT can also be seen in other conditions that create a myocardial scar, such as the following (see Etiology):
-
Dilated cardiomyopathy
-
Hypertrophic cardiomyopathy
-
Arrhythmogenic right ventricular dysplasia (ARVD) or cardiomyopathy
-
Chagas disease [10]
-
Surgical incisions in the ventricle
VT may also occur in the absence of structural heart disease. VT in this setting may result from enhanced automaticity, which most commonly originates in the right ventricular outflow tract or from the fascicles of the cardiac conduction system. Bundle-branch reentrant VT occurs in patients with conduction system disease distal to the bundle of His. Finally, functional reentrant VTs can occur in structurally normal hearts, in patients with inherited channelopathies. [11] The VT morphology can provide a guide to the anatomic likely site of origin in the heart. [12, 13]
VT can also be triggered by the following (see Etiology):
-
Systemic diseases that affect the myocardium (eg, sarcoidosis, amyloidosis, systemic lupus erythematosus, hemochromatosis, rheumatoid arthritis)
-
Sympathomimetic agents, including intravenous (IV) inotropes and illicit drugs such as methamphetamine or cocaine
-
Digitalis toxicity, which can lead to biventricular tachycardia
-
Drugs that prolong the QT interval (class IA and class III antiarrhythmics, azithromycin, levofloxacin, and many others); these may cause torsade de pointes
-
Drugs that slow conduction velocity, particularly when an underlying myocardial scar is present (eg, halothane and class IA and IC antiarrhythmics)
Clinically, VT may be reflected in symptoms such as syncope, palpitations, and dyspnea (see Presentation). It is often, but not always, associated with hemodynamic compromise, particularly if the left ventricle is impaired or the heart rate is especially fast. With some exceptions, VT is associated with increased risk of sudden death. [1, 14] (See Prognosis.)
The ECG diagnosis of VT is generally straightforward, but it does require that this condition be distinguished from aberrantly conducted supraventricular tachycardia (SVT), which has a similar ECG pattern. ECG criteria for confirming the presence of a VT mechanism for a wide-complex tachycardia include the following:
-
Presence of atrioventricular (AV)—technically, ventriculoatrial—dissociation (in which the ventricles fire at a faster rate than the atria)
-
Fusion beats
-
Capture beats
Because AV dissociation, fusion, and capture beats occur in only a minority of VT tracings, additional 12-lead ECG criteria (the Brugada criteria [15] and the Vereckei criteria [16] ) have been derived to facilitate discrimination between VT and aberrantly conducted SVT. (See Workup).
Accelerated idioventricular rhythm, sometimes termed slow VT, is a variant of VT that produces a rate of 60-120 beats/min. It typically occurs in patients with underlying heart disease (ischemic or structural), is transient, and only rarely is associated with hemodynamic compromise or collapse. Treatment of the dysrhythmia itself usually is not required unless significant hemodynamic impairment develops.
Patients with frank hemodynamic compromise from acute VT require emergency management with electrical cardioversion. Although cardioversion treats VT, it does not prevent recurrence of VT, and patients may experience repeated episodes of recurrent VT after cardioversion; this phenomenon is termed VT storm. These patients additionally require acute antiarrhythmic therapy, ablation therapy, or both.
The mainstays of long-term treatment for clinically stable patients with VT are the various antiarrhythmic drugs. However, cardiologists are increasingly making use of interventional therapy with devices and ablation procedures designed to abort VT or to destroy arrhythmogenic tissue in the heart. (See Treatment.)
For information on VT in children, see Pediatric Ventricular Tachycardia. For patient education information, see the Heart Health Center, as well as Arrhythmias (Heart Rhythm Disorders), Supraventricular Tachycardia (SVT, PSVT), and Palpitations.
Pathophysiology
At the cellular level, ventricular tachycardia (VT) is caused by electrical reentry or abnormal automaticity. Myocardial scarring from any process increases the likelihood of electrical reentrant circuits. These circuits generally include a zone where normal electrical propagation is slowed by the scar. Ventricular scar formation from a prior myocardial infarction (MI) is the most common cause of sustained monomorphic VT.
VT in a structurally normal heart typically results from mechanisms such as triggered activity and enhanced automaticity. Torsade de pointes, seen in the long QT syndromes, is likely a combination of triggered activity and ventricular reentry. [17]
During VT, cardiac output is reduced as a consequence of decreased ventricular filling from the rapid heart rate and the lack of properly timed or coordinated atrial contraction. Ischemia and mitral insufficiency [18] may also contribute to decreased ventricular stroke output and hemodynamic intolerance.
Hemodynamic collapse is more likely when underlying left ventricular dysfunction is present or when heart rates are very rapid. Diminished cardiac output may result in diminished myocardial perfusion, worsening inotropic response, and degeneration to ventricular fibrillation (VF), resulting in sudden death.
In patients with monomorphic VT, mortality risk correlates with the degree of structural heart disease. Underlying structural heart diseases such as ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, Chagas disease, and right ventricular dysplasia have all been associated with degeneration of monomorphic or polymorphic VT to VF. [10] Even without such degeneration, VT can also produce congestive heart failure and hemodynamic compromise, with subsequent morbidity and mortality.
If VT is hemodynamically tolerated, the incessant tachyarrhythmia may cause a dilated cardiomyopathy. This may develop over a period of weeks to years and may resolve with successful suppression of the VT. [19] A similar course is occasionally seen in patients with frequent premature ventricular contractions or ventricular bigeminy, despite the absence of sustained high rates. [20]
Etiology
Causes of ventricular tachycardia (VT) include the following [11] :
-
Ischemic heart disease (most common)
-
Structural heart disease with disruption of normal conduction patterns (eg, nonischemic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy [ARVD] or cardiomyopathy, hypertrophic cardiomyopathy)
-
Congenital structural cardiac disorders (eg, tetralogy of Fallot) and associated surgical scar
-
Acquired channelopathies, most commonly from drugs that prolong the QT interval (eg, class IA and class III antiarrhythmics, phenothiazines, methadone, many others); drugs that slow myocardial conduction (eg, flecainide, propafenone, halothane) may also promote reentrant VT
-
Inherited channelopathies (eg, long QT syndrome, short QT syndrome, Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia)
-
Sympathomimetic agents, including intravenous (IV) inotropes and illicit drugs such as methamphetamine or cocaine
-
Digitalis toxicity, which can lead to biventricular tachycardia
-
Systemic diseases causing infiltrative cardiomyopathy or scar (eg, sarcoidosis, amyloidosis, systemic lupus erythematosus, hemochromatosis, rheumatoid arthritis)
Hypokalemia is an important arrhythmia trigger, followed by hypomagnesemia. Hyperkalemia may also predispose to VT and ventricular fibrillation (VF), particularly in patients with structural heart disease. Other triggers include sleep apnea and atrial fibrillation (AF), which can increase VT risk in patients with preexisting structural heart disease.
QT prolongation, which may be acquired or inherited, can lead to VT. Acquired QT prolongation is observed with certain potassium channel–blocking medications. Most of the causative drugs block the delayed rectifier cardiac potassium current, IKr. These agents include class IA and class III antiarrhythmics, azithromycin, and many others. Congenital long QT syndrome is a group of genetic disorders involving abnormal cardiac ion channels (most commonly, potassium channels responsible for ventricular repolarization).
In both acquired and congenital long QT syndromes, prolonged repolarization predisposes to torsade de pointes, a reentrant rhythm with a constantly varying circuit. [17] Other inherited ion channel abnormalities may cause idiopathic VF and familial polymorphic VT in the absence of QT prolongation.
Although the following syndromes have been described in most parts of the world, population groups in certain regions carry locally increased risk of genetically mediated heart disease. Examples include the Veneto region of Italy and the Greek island of Naxos (right ventricular dysplasia), [21] as well as northeastern Thailand (idiopathic VF/Brugada syndrome). [22] The risk for VT within populations varies primarily with the risk factors for atherosclerosis, however, rather than with ethnic differences per se.
Among patients younger than 35 years, the most common cardiac causes of sudden death, and presumably of VT, include the following [23] :
-
Hypertrophic cardiomyopathy
-
Right ventricular cardiomyopathy (ARVD)
-
Long QT syndrome
-
Congenital coronary artery abnormalities
Inherited long QT syndrome
Long QT syndrome is characterized by QT interval prolongation, T-wave abnormalities, and polymorphic VT. Persons with this syndrome are predisposed to episodes of polymorphic VT. These episodes can be self-limited, resulting in syncope, or they may transition into VF and thus can cause sudden cardiac death.
Long QT syndromes have been identified by eponyms (ie, Romano-Ward syndrome, Jervell and Lange-Nielsen syndrome, Andersen-Tawil syndrome, [24] and Timothy syndrome [25] ). The form sometimes known as Romano-Ward syndrome is the most common type. However, current practice is moving away from using eponyms and toward denoting these syndromes as numbered types (eg, LQT1 through LGT12) on the basis of identified underlying mutations.
Mutations in the KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 genes are known to be causative. Together, those five genes are responsible for virtually 100% of cases of inherited long QT syndrome.
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic VT (CPVT) is characterized by polymorphic VT that can be triggered by stress, exercise, or even strong emotional states. It can be induced by catecholamine administration. Patients may present with syncope or with sudden cardiac death if the dysrhythmia degrades into VF. Physical examination or electrocardiography (ECG) during rest will likely be normal.
CPVT may be caused by mutations in the CASQ2 or RYR2 genes. [26] An additional locus has been mapped to chromosome 7p22-p14. This disorder shares clinical characteristics with the bidirectional VT sometimes seen in digitalis toxicity.
Dilated cardiomyopathy
Dilated cardiomyopathy is a highly heterogeneous disorder that can predispose to ventricular tachyarrhythmias such as VT. Its genetic causes are myriad and involve mutations in genes coding for proteins that make up cardiac sarcomeres, including actin, myosin, and troponin. It is noteworthy that genes such as PSEN1 and PSEN2, which are responsible for early-onset Alzheimer disease, have also been implicated in dilated cardiomyopathy.
Most familial dilated cardiomyopathies are inherited in an autosomal dominant fashion. X-linked inheritance of dilated cardiomyopathy has been described in patients with mutations in the DMD gene (Duchenne muscular dystrophy) and the TAZ gene (Barth syndrome). Autosomal recessive inheritance has been described in mutations of the TNNI3 gene, which encodes troponin I.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy is usually inherited in an autosomal dominant fashion with incomplete penetrance. Mutations in four genes that encode sarcomeric proteins—TNNT2, MYBPC3, MYH7, and TNNI3—account for approximately 90% of cases. [27] Most people with symptomatic hypertrophic cardiomyopathy will experience them at rest. Less often, a person with this disorder will experience an initial episode of VT or VF with significant exertion.
Arrhythmogenic right ventricular dysplasia
ARVD (also known as right ventricular cardiomyopathy) is characterized by replacement of the free wall of the right ventricle with fibrous tissue and the development of right ventricular hypertrophy. This disorder frequently results in sustained VT, which may degrade into VF.
The genetics of ARVD are extremely heterogeneous. More than 10 genes (eg, TGFB3, RYR2, DSP, PKP2, DSG2, DSC2, [28] TMEM43, JUP [29] ) and seven additional loci (eg, 14q12-q22, 2q32.1-32.3, 10p14-p12, 10q22) have been implicated in the pathogenesis of this disorder, which is inherited in an autosomal dominant fashion with incomplete penetrance. [29] Those genes are believed to be responsible for approximately 40-50% of the total cases of ARVD. [28]
Brugada syndrome
Brugada syndrome is characterized by the specific ECG pattern of right bundle-branch block and ST-segment elevation in the early precordial leads, most commonly V1-V3, without any structural abnormality of the heart. It causes idiopathic VT or VF and carries a high risk for sudden cardiac death. [30]
Brugada syndrome can be caused by many genes. At least nine genes are known to cause Brugada syndrome (SCN5A, GPD1L, CACNA1C, CACNB2, SCN1B, KCNE3, SCN3B, HCN4, and KCND3), but SCN5A accounts for about 20% of cases, with other known "minor" genes comprising another 15% of cases. [31] Brugada syndrome is inherited in an autosomal dominant fashion.
Familial ventricular tachycardia
Familial VT is characterized by paroxysmal VT in the absence of cardiomyopathy or another identifiable electrophysiologic disorder. Familial VT is rare; investigation of families with paroxysmal VT will frequently reveal disorders such as Brugada syndrome, long QT syndrome, or catecholaminergic polymorphic VT. In at least one case, however, these disorders were ruled out, and the patient was found to have a somatic mutation in the GNAI2 gene. [32]
Epidemiology
Ventricular tachycardia (VT) and coronary artery disease (CAD) are common throughout most of the developed world. In developing countries, VT and other heart diseases are relatively less common.
The incidence of VT in the United States is not well quantified, because of the clinical overlap of VT with ventricular fibrillation (VF), but examination of sudden death data provides a rough estimate of VT incidence. Most sudden cardiac deaths are caused by VT or VF, [1] at an estimated rate of approximately 300,000 deaths per year in the United States, or about half of the estimated cardiac mortality. [2]
A prospective surveillance study gave a sudden death incidence of 53 per 100,000 population, accounting for 5.6% of all mortality. [33] This is only a rough estimate of VT incidence, both because many patients have nonfatal VT and because arrhythmic sudden deaths may be associated with VF or bradycardia rather than with VT. In patients with ischemic cardiomyopathy and nonsustained VT, sudden death mortality approaches 30% in 2 years.
Morbidity from VT is associated with hemodynamic collapse. Resuscitated survivors may suffer ischemic encephalopathy, acute renal insufficiency, transient ventricular dysfunction, aspiration pneumonitis, and trauma related to resuscitative efforts.
Age-related demographics
VT is unusual in children but may occur in the postoperative cardiac setting or in patients with associated congenital heart disease. Tachydysrhythmias in children are more commonly due to paroxysmal supraventricular tachycardias (PSVTs). [34] The incidence of ischemic VT increases with age, regardless of sex, as the prevalence of CAD increases. VT rates peak in the middle decades of life, in concert with the incidence of structural heart disease. Idiopathic VT can be observed at any age.
Sex-related demographics
VT is observed more frequently in men because ischemic heart disease is more prevalent in men. Among patients with CAD in the Framingham Heart Study, male deaths were more common than female deaths (46% vs 34%, respectively). [35] It seems certain that as CAD becomes more common in women, the incidence of VT in women will increase.
Females with acquired or congenital long QT syndromes are at greater risk for sudden death. The opposite is true for arrhythmogenic right ventricular cardiomyopathy (a two-fold male predominance) and Brugada syndrome (an approximately eight-fold male predominance).
Prognosis
The prognosis in patients with ventricular tachycardia (VT) varies with the specific cardiac process, but it is predicted best by left ventricular function. Patients with VT may suffer heart failure and its attendant morbidity as a result of hemodynamic compromise. In patients with ischemic cardiomyopathy and nonsustained VT, sudden-death mortality approaches 30% in 2 years. In patients with idiopathic VT, the prognosis is excellent, with the major risk being injury incurred during syncopal spells.
Data from the Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction Trial suggest that VT or ventricular fibrillation occurring before coronary angiography and revascularization in the setting of ST-segment elevation myocardial infarction has a strong association with increased 3-year rates of death and stent thrombosis. [36]
Appropriate treatment can significantly improve the prognosis in selected patients. Beta-blocker therapy can reduce the risk of sudden cardiac death from VT, and implantable cardioverter-defibrillators can terminate malignant arrhythmias. [37]
The prognosis does not always correlate with left ventricular function. Patients with long QT syndrome, right ventricular dysplasia, or hypertrophic cardiomyopathy may be at increased risk for sudden death despite relatively well preserved left ventricular function. These possibilities should be considered in any patient with a strong family history of premature sudden death.
-
This electrocardiogram (ECG) shows rapid monomorphic ventricular tachycardia (VT), 280 beats/min, associated with hemodynamic collapse. The tracing was obtained from a patient with severe ischemic cardiomyopathy during an electrophysiologic study. A single external shock subsequently converted VT to sinus rhythm. The patient had an atrial rate of 72 beats/min (measured with intracardiac electrodes; not shown). Although ventriculoatrial dissociation (faster V rate than A rate) is diagnostic of VT, surface ECG findings (dissociated P waves, fusion or capture beats) are present in only about 20% of cases. In this tracing, the ventricular rate is simply too fast for P waves to be observed. VT at 240-300 beats/min is often termed ventricular flutter.
-
This electrocardiogram shows slow monomorphic ventricular tachycardia (VT), 121 beats/min, from a patient with an old inferior wall myocardial infarction and well-preserved left ventricular (LV) function (ejection fraction, 55%). The patient presented with symptoms of palpitation and neck fullness. Note the ventriculoatrial dissociation, which is most obvious in leads V2 and V3. Slower VT rates and preserved LV function are associated with better long-term prognosis.
-
At first glance, this tracing suggests rapid polymorphic ventricular tachycardia. It is actually sinus rhythm with premature atrial complex and a superimposed lead motion artifact. Hidden sinus beats can be observed by using calipers to march backward from the final two QRS complexes. This artifact can be generated easily with rapid arm motion (eg, brushing teeth) during telemetry monitoring.
-
Torsade de pointes. Image A: This is polymorphic ventricular tachycardia associated with resting QT-interval prolongation. In this case, it was caused by the class III antiarrhythmic agent sotalol. This rhythm is also observed in families with mutations affecting certain cardiac ion channels. Image B: Torsade de pointes, a form of ventricular tachycardia. Courtesy of Science Source/BSIP.
-
Preexcited atrial fibrillation. The patient has an accessory atrioventricular connection. Atrial fibrillation has been induced. Conduction over an accessory pathway results in a wide QRS complex, mimicking ventricular tachycardia.
-
Curative ablation of ventricular tachycardia (VT). The patient had VT in the setting of ischemic cardiomyopathy. VT was induced in an electrophysiology laboratory, and an ablation catheter was placed at the critical zone of slow conduction within the VT circuit. Radiofrequency (RF) energy was applied to tissue through the catheter tip, and VT was terminated when the critical conducting tissue was destroyed.
-
Ventricular pacing at 120 beats/min. Newer pacemakers use bipolar pacing. If a smaller pacing stimulus artifact is overlooked, an erroneous diagnosis of ventricular tachycardia may result. Because leads are most commonly placed in the right ventricular apex, paced beats will have a left bundle-branch block morphology with inferior axis. Causes of rapid pacing include (1) tracking of atrial tachycardia in DDD mode, (2) rapid pacing due to the rate response being activated, and (3) endless loop tachycardia. Application of a magnet to the pacemaker will disable sensing and allow further diagnosis. Sometimes “pacing spike detection” must be programmed “ON” in the electrocardiographic system to make the spike apparent.
-
Supraventricular tachycardia with aberrancy. This tracing is from a patient with a structurally normal heart who has a normal resting electrocardiogram. This rhythm is orthodromic reciprocating tachycardia with rate-related left bundle-branch block. Note the relatively narrow RS intervals in the precordial leads.
-
Termination of ventricular tachycardia (VT) with overdrive pacing. This patient has reentrant VT, which is terminated automatically by pacing from an implantable cardioverter-defibrillator.
-
Posteroanterior view of a right ventricular endocardial activation map during ventricular tachycardia in a patient with a previous septal myocardial infarction. The earliest activation is recorded in red, and late activation as blue to magenta. Fragmented low-amplitude diastolic local electrograms were recorded adjacent to the earliest (red) breakout area, and local ablation in this scarred zone (red dots) resulted in termination and noninducibility of this previously incessant arrhythmia.
-
This tracing depicts monomorphic ventricular tachycardia.
-
This image demonstrates polymorphic ventricular tachycardia.
-
This electrocardiogram is from a 32-year-old woman with recent-onset heart failure and syncope.
-
This electrocardiogram is from a 48-year-old man with wide-complex tachycardia during a treadmill stress test. Any wide-complex tachycardia tracing should raise the possibility of ventricular tachycardia, but closer scrutiny confirms left bundle-branch block conduction of a supraventricular rhythm. By Brugada criteria, RS complexes are apparent in the precordium (V2-V4), and the interval from R-wave onset to the deepest part of the S wave is shorter than 100 ms in each of these leads. Ventriculoatrial dissociation is not seen. Vereckei criteria are based solely upon lead aVR, which shows no R wave, an initial q wave width shorter than 40 ms, and no initial notching in the q wave. The last Vereckei criterion examines the slope of the initial 40 ms of the QRS versus the terminal 40 ms of the QRS complex in lead aVR. In this case, the initial downward deflection in lead aVR is steeper than the terminal upward deflection, yielding Vi/Vt ratio above 1. All of these criteria are consistent with an aberrantly conducted supraventricular tachycardia. Gradual rate changes during this patient's treadmill study (not shown here) were consistent with a sinus tachycardia mechanism.
-
The electrocardiogram shows a form of idiopathic ventricular tachycardia (VT) seen in the absence of structural heart disease. This rhythm arises from the left ventricular septum and often responds to verapamil. Upon superficial examination, it appears to be supraventricular tachycardia with bifascicular conduction block. Closer examination of lead V1 shows narrowing of fourth QRS complex, consistent with fusion between the wide QRS complex and the conducted atrial beat, confirming atrioventricular dissociation and a VT mechanism.
-
A wide QRS complex tachycardia is evident on this electrocardiogram from a 64-year-old man with history of previous myocardial infarction (MI) and syncope. In patients with a prior MI, the most common mechanism of wide QRS complex tachycardia is ventricular tachycardia.
-
This tracing depicts atrioventricular dissociation.
-
Fusion beats, capture beats, and atrioventricular dissociation can be seen on this electrocardiogram.
-
Note the retrograde P waves in this electrocardiogram.
-
Retrograde P waves are also observed in this electrocardiogram.
-
This electrocardiogram reveals torsade de pointes.
-
Hematoxylin and eosin stain; intermediate power of a healed myocardial infarct. Note the areas of fibrosis (pale pink) dissecting between the myocytes (red).
Tables
| Recommendation | Class |
|---|---|
| Resting 12-lead electrocardiography (ECG) in all patients | Class I |
12-lead ambulatory ECG to evaluate QT-interval changes or ST changes |
Class I |
| Cardiac event recorders when symptoms are sporadic to rule out transient arrhythmias | Class I |
Implantable loop recorders when symptoms are sporadic and suspected to be related to arrhythmias and when a symptom–rhythm correlation cannot be established by conventional diagnostic techniques |
Class I |
Exercise stress testing in adult patients who have an intermediate or greater probability of having coronary artery disease (CAD) to provoke ischemic changes or ventricular arrhythmia (VA) |
Class I |
| Exercise stress testing in patients with known or suspected exercise-induced VA | Class I |
| Echocardiography in all patients | Class I |
Pharmacologic stress testing plus imaging modality study to detect silent ischemia in patients with VAs who have an intermediate probability of having CAD and are physically unable to perform a symptom-limited exercise test |
Class I |
Cardiac magnetic resonance imaging (cMRI) or computed tomography (CT) scanning in patients with VAs when echocardiography does not provide accurate assessment of left- and right-ventricular function and/or evaluation of structural changes |
Class IIa |
Electrophysiologic study in patients with CAD with remote myocardial infarction with symptoms suggestive of ventricular tachyarrhythmias, including palpitations, presyncope, and syncope. |
Class I |
Coronary angiography to establish or exclude significant obstructive CAD in patients with life-threatening VAs or in survivors of sudden cardiac death, who have an intermediate or greater probability of having CAD by age and symptoms |
Class IIa |





