Practice Essentials
Antidysrhythmic medications are widely used to treat or prevent abnormalities in cardiac rhythms. They accomplish this through a number of mechanisms involving automaticity or ion channel dynamics, which in turn affect the propagation of the myocardial electrical impulse via change in conduction velocity or refractory period.
Antidysrhythmics alter the propagation and mechanisms of cardiac rhythms, making toxicity from these agents highly lethal. In fact, antidysrythmics can be prodysrhythmic at both therapeutic and toxic serum concentrations. Additionally, the patients receiving these drugs may have a lower dysrhythmic threshold resulting from underlying cardiac conditions as well as other comorbidities, making them more suscpetible to toxicity. A thorough knowledge of this class of drugs is necessary for differentiating drug toxicity from primary disease.
See also Beta-Blocker Toxicity and Calcium Channel Blocker Toxicity, as those topics are not covered in this article.
Signs and symptoms
Toxicity from antidysrhythmic agents can be grouped in terms of clinical presentation and electrocardiographic (ECG) abnormalities, as follows:
-
Anticholinergic syndromes: Procainamide, disopyramide, quinidine
-
Heart failure: Disopyramide, flecainide, propafenone, sotalol, procainamide
-
Hypotension: Quinidine, procainamide, amiodarone, dronedarone, lidocaine, mexiletine, flecainide, propafenone, ibutilide
-
CNS symptoms: Quinidine, procainamide, disopyramide, lidocaine, mexiletine, flecainide, propafenone
-
Seizures (acute toxicity): Quinidine, procainamide, lidocaine, mexiletine, flecainide, propafenone
-
Endocrine dysfunction: Amiodarone (hypothyroidism or hyperthyroidism), quinidine and disopyramide (hypoglycemia)
-
Hematology/Oncology: Amiodarone (hepatic/biliary malignancy), procainamide (blood dyscrasia)
-
Pulmonary/autoimmune: Procainamide (lupuslike syndrome, vasculitis), amiodorone (fibrosis, pneumonitis)
-
CNS: procainamide, lidocaine, mexilitine
-
Dermatologic: procainamide, propafenone
ECG changes are as follows:
-
QRS widening: Quinidine, procainamide, disopyramide, flecainide, propafenone, amiodarone, dronedarone
-
PR prolongation: procainamide
-
QTc prolongation: Quinidine, procainamide, disopyramide, amiodarone, dronedarone, sotalol, ibutilide, dofetilide; slight prolongation with flecainide, propafenone
-
Ventricular arrythmia (ventricular premature contractions or ventricular tachycardia): Procainamide
-
Torsade de pointes: Ibutilide, dofetilide, sotalol, quinidine, procainamide, disopyramide
See Clinical Presentation for more detail.
Diagnosis
The first and most important diagnostic tool in acute antidysrhythmic toxicity is electrocardiography. ECG changes such as QRS widening, QTc prolongation, and atrioventricular block should be ruled out.
Serum electrolytes concentrations should be obtained, especially in patients taking antidysrhythmics that prolong the correct QT (QTc) interval.
Serum drug concentrations are not likely to be helpful to the emergency physician treating a patient with acute antidysrhythmic drug toxicity, but concentrations of quinidine, lidocaine, and propafenone can be measured in the acute care setting.
Chest radiographs and brain natriuretic peptide levels should be obtained in patients presenting with heart failure symptoms; Chest radiographs should also be obtained in patients taking amiodarone or dronedarone and presenting with pulmonary symptoms.
Thyroid function tests should be obtained in patients taking amiodarone or dronedarone who present with signs and symptoms of hypothyroidism or hyperthyroidism.
See Workup for more detail.
Management
Airway, breathing, and circulatory support; intravenous access; and ECG monitoring are of paramount importance. Treatment measures and the drugs for which they are appropriate are as follows:
-
GI decontamination: Should be considered in most antidyrthymmic overdose, and specifically with disopyramide, quinidine, flecainide, propafenone, amiodarone
-
Hemodialysis: Procainamide, mexiletine, sotalol
-
Sodium bicarbonate: Class Ia and Ic antidysrhythmics (quinidine, procainamide, disopyramide, lidocaine, mexiletine, flecainide, propafenone)
-
Magnesium: Class III agents, Ibutilide, dofetilide, sotalol, quinidine, procainamide, disopyramide
-
Intravenous lipid emulsion: Verapamil; less evidence for lidocaine, flecainide, amiodarone
-
Seizure control (benzodiazepines): Quinidine, procainamide, lidocaine, mexiletine, flecainide, propafenone
See Treatment and Medication for more detail.
Background
Despite the advent of interventional techniques such as catheter ablation and the implantable cardioverter-defibrillator in the treatment of supraventricular and ventricular tachycardia, antidysrhythmic drugs continue to play a significant role in treating and suppressing life-threatening dysrhythmias. The prodysrhythmic effects of many of these drugs also continue to present a major clinical problem, especially in the growing population of patients with underlying heart failure.
When encountering a patient with dysrhythmias on antidysythmic drugs, the physician must maintain a broad differential diagnosis that includes not only drug toxicity but underlying ischemia, structural cardiac abnormalities, and conduction disturbances. Thus, understanding the adverse effects and electrocardiographic profiles of antidysrhythmic agents is critical for diagnosis and treatment of possibly life-threatening drug toxicity.
This article discusses the major antidysrhythmic drugs within classes I, III and V, with specific attention to their adverse effects and clinical presentations in the setting of acute toxicity. Toxicity from class II and IV dysrhythmics is discussed elsewhere (see Beta-Blocker Toxicity and Calcium Channel Blocker Toxicity)
For additional information, see Medscape's Cardiology Resource Center. For patient education resources, see the First Aid and Injuries Center, as well as Poisoning, Drug Overdose, Activated Charcoal, and Poison Proofing Your Home.
Pathophysiology
Most antidysrythmics may be categorized via the Vaughan-Williams classification system, based on their mechanism of activity (see the image below). Medications used to treat arrhythmias that have variable mechanisms have been included in class V; these include magnesium, digoxin, and adenosine. The Vaughn-Williams classes are as follows:
-
Class I: Sodium channel blockers
-
Class II: Beta-adrenergic blockers
-
Class III: Potassium channel blockers
-
Class IV: Calcium channel blockers
-
Class V: Other or unknown mechanism of action
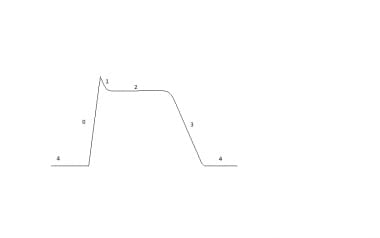 Schematic of the cardiac action potential. Phase 0 depicts the the influx of sodium ions. Phases 1 and 3 correspond to the sodium-channel inactivation and the repolarizing eflux of potassium ions, respectively. Phase 2 depicts the opening of voltage-sensitive calcium channels causing a plateau in voltage.
Schematic of the cardiac action potential. Phase 0 depicts the the influx of sodium ions. Phases 1 and 3 correspond to the sodium-channel inactivation and the repolarizing eflux of potassium ions, respectively. Phase 2 depicts the opening of voltage-sensitive calcium channels causing a plateau in voltage.
Class I agents bind sodium channels reducing depolarization rate, which serves to slow and reduce the rate of rise of the action potential (phase 0). They also help to inhibit depolarization of neuronal cells, which provides local anesthesia. Class I agents also inhibit depolarization in atrial, ventricular, and Purkinje myocytes, thereby decreasing conduction velocity and automaticity.
Class I agents are further categorized into A, B, or C subclasses, based on the degree of sodium channel blockade and effects on repolarization, as follows:
-
Class IA agents: Prolong repolarization and action potential through blockade of outward rectifying potassium channels; moderate slowing of cardiac conduction, prolong both the QRS and QTc intervals
-
Class IB agents: Bind to the sodium channel in its inactivated state; shorten action potential duration and selectively depress cardiac conduction in ischemic cells; generally do not prolong the QRS interval
-
Class IC agents: Bind to sodium channels in the active state and are slow to release from sodium channels; decrease rate of rise of phase 0 of the action potential leading to prolonged QRS interval; have little effect on action potential duration but markedly depress cardiac conduction
Class II agents indirectly blockade calcium channel opening by attenuating adrenergic activation. These agents block the proarrhythmic effects of catecholamines.
Class III agents prolong refractoriness and delay repolarization by blocking potassium channels (phase 2, phase 3) leading to prolonged QTc intervals on the ECG. They have little direct effect on sodium channels.
Class IV agents slow sinoatrial node pacemaker cell and atrioventricular conduction by direct blockade of L-type voltage-gated calcium channels.
Etiology
Class IA antidysrhythmics
Disopyramide
In addition to sodium and potassium channel blockade, disopyramide is a muscarinic antagonist. See the following:
-
Indications: Documented ventricular dyshythmias, atrial dysrhythmias in patients with hypertrophic cardiomyopathy (unlabeled use)
-
Dosages: Dose adjustment is gradual; 100-200 mg orally every 6 hours; reduced dosage frequency recommended in renally impaired patients
-
Metabolism: Metabolized by the liver (CYP3A4), 40-60% excreted by the kidneys
-
Therapeutic concentrations: Atrial dysrhythmias at 2.8-3.2 mcg/mL, ventricular dysrhythmias at 3.3-7.5 mcg/mL, toxic level at >7 mcg/mL
-
Drug interactions: Drugs that also prolong the QTc interval should be avoided, as torsade de pointes can occur with coadministration; drugs that interfere with CYP3A4 hepatic metabolism of disopyramide include ketoconazole, isoniazid, erythromycin, phenytoin, rifampin, verapamil and protease inhibitors used for HIV infection; disopyramide may enhance anticholinergic effects of anticholinergic agents, such as dry mouth, urinary retention, and constipation. Disopyramide also possesses strong negative inotropic activity and should be avoided in those with left ventricular dysfunction. as well as those taking β-blockers. [1]
Procainamide
Procainamide blocks sodium and potassium channels, and its active metabolite prolongs the action potential duration of ventricular myocytes and Purkinje fibers. It is available in oral, intramuscular (IM), and intravenous (IV) forms. See the following:
-
Indications: Supraventricular or ventricular dysrhythmias
-
Dosages - Loading dose IV: 15-18 mg/kg over 30 minutes IV or 20-50 mg/minute or 100 mg every 5 minutes until dysrhythmia controlled, QRS widens to 50% its original width, hypotension occurs, or maximum of 17 mg/kg has been given; IV maintenance dose is 1-4 mg/min; IM: 0.5-1 g every 4-8 hours; toxicity may occur if dose is not reduced in patients with renal or hepatic impairment; oral form is not available in the United States
-
Metabolism: Metabolized in the liver by acetylation into a metabolite that prolongs the action potential; both procainamide and its metabolite are excreted by the kidneys
-
Therapeutic concentrations: 4-10 µg/mL; toxic concentration at >10-12 µg/mL; severe toxicity when serum concentrations >60 µg/mL [2]
-
Drug interactions: Anticholinergic drugs produce additive vagolytic effects; QTc prolongation can become more severe when patients are taking other QTc-prolonging drugs; cimetidine increases drug levels of procainamide by interfering with its hepatic metabolism
Procainamide should be avoided in patients with myasthenia gravis.
Quinidine
In addition to blocking sodium and potassium channels, quinidine blocks alpha-adrenergic receptors and muscarinic receptors. Quinidine has the same antimalarial and antipyretic properties as quinine; in addition to its cardiologic indications, it is used for treatment of malaria, and as an illicit abortifacient. See the following:
-
Indications: Suppression of atrial and ventricular dysrhythmias; quinidine has shown effective antiarrhythmic activity in patients with Brugada syndrome [3]
-
Test dose: Test dose of 200mg quinidine sulfate several hours before full dosage
-
Maintenance dose: quinidine sulfate 200-400mg PO q6-8hr or 600mg of SR PO q8-12hr
-
Maintenance dose: quinidine gluconate 648mg PO q12hr OR 324-660mg PO q8hr
-
Metabolism: Hepatic elimination is responsible for 60-80%, whereas renal elimination is responsible for 20-40%
-
Therapeutic concentrations: 2-6 mg/L or 6.2-18.5 µmol/L by assay; concentrations >14 µg/mL are associated with toxicity
-
Drug interactions: Cimetidine and ketoconazole elevate the quinidine serum concentration; verapamil impairs hepatic metabolism, quinidine increases digoxin concentration
Risk factors for quinidine toxicity are hepatic disease, renal insufficiency, and heart failure. Quinidine can cause sinus node depression in patients with sick sinus syndrome.
Class IB antidysrhythmics
Lidocaine
Lidocaine is a derivative of cocaine that blocks fast sodium channels, leading to a modest reduction in the rate of phase 0 depolarization. See the following:
-
Indications: Ventricular dysrhythmias; local anesthetic; no longer used to prevent dysrhythmias immediately following myocardial infarction, with amiodarone being the preferred agent [4]
-
Dosages: Initial bolus of 1 mg/kg at a rate of 20-50 mg/min, then 0.5 mg/kg, 20-40 minutes later; maintenance infusion rate of 1-4 mg/min
-
Metabolism: Hepatic metabolism by CYP3A4 to an active metabolite; toxicity more likely to occur in patients with reduced hepatic blood flow (eg, from shock, low cardiac output) or hepatic dysfunction (eg, cirrhosis)
-
Therapeutic concentrations: Toxic concentrations at >5 µg/mL; severe toxicity when concentrations >10 µg/mL
-
Drug interactions: CYP3A4 inhibitors such as cimetidine and amiodarone increase risk of lidocaine toxicity; beta-blockers, especially nonselective ones such as propranolol, may reduce hepatic blood flow, leading to decreased clearance of lidocaine and increased drug effect
The therapeutic index of lidocaine is narrow. Toxicity may occur while a clinician is trying to achieve adequate local or regional anesthesia for repairing large lacerations or from pediatric ingestion of viscous lidocaine. Patients at greatest risk for iatrogenic toxicity are those with poor cardiac output or hepatic disease. Toxicity is potentiated in acidemic states (eg, hypercapnia during rapid sequence intubation, lactic acidosis following seizure). [5]
Mexiletine
Mexiletine is clinically equivalent to lidocaine in its mechanism of slowing the rate of phase 0 depolarization by blocking fast sodium channels, and it shortens the action potential duration of Purkinje fibers. It blocks the late sodium current, which may be useful for preventing delayed ventricular repolarization and torsade de pointes in long QT syndrome. [6]
-
Indications: Ventricular dyshythmias, neuropathic pain, myotonia
-
Dosages: 200-300 mg every 6-8 hours; dosage should be reduced in patients with hepatic dysfunction or heart failure due to decreased clearance
-
Metabolism: Rapidly absorbed by the small intestine and undergoes hepatic, primarily CYP2D6, metabolism; patients with decreased hepatic blood flow are at increased risk for toxicity; 10% of mexiletine is eliminated unchanged by the kidneys; urine alkalization slows renal elimination
-
Therapeutic concentrations: 0.5-2 µg/mL; toxic concentration at >2 µg/mL
-
Drug interactions: Cimetidine increases risk of toxicity. Selective serotonin reuptake inhibitors may decrease mexiletine clearance and promote toxicity. [7]
Class IC antidysrhythmics
Flecainide
Flecainide has a strong blocking effect on the rapid sodium channel, decreasing the rate of depolarization. Flecainide also slows conduction in all cardiac fibers, making it contraindicated in patients with second degree atrioventricular block and intraventricular conduction delay. In high concentrations, flecainide may block slow calcium channels and have negative inotropic effects. See the following:
-
Indications: Supraventricular dysrhythmias (primarily paroxysmal atrial fibrillation) and ventricular dysrhythmias; however, was shown to increase dysythymias and mortality in the post-MI period and those with depressed LV function and history of ventriclar dysrythmias; also used to help diagnose or possibly treat certain congential dysryhtmia disorders such as Brugada and LQT3 syndrome. [8, 9]
-
Dosages: Starting dose 100 mg every 12 hours, not to exceed 400 mg/day; some recommend initiating therapy as an inpatient to monitor for dysrhythmias
-
Metabolism: 75% undergoes hepatic CYP2D6 metabolism; 25% is renally eliminated
-
Therapeutic concentrations: 0.2-1 mg/mL
-
Drug interactions: May increase serum concentrations of digoxin and propranolol; drugs such as paroxetine, fluoxetine, quinidine, amiodarone, propranolol, and ritonavir can increase flecainide concentrations by affecting CYP2D6 metabolism. Should be avoided with beta blockers due to concomitant AV nodal suppression and negative inotropy. Concomitant thiazide and flecainaide use predisposes patients to hyponatremia which can inturn precipitate flecainaide toxicity.
Patients at risk for flecainide toxicity include those with renal insufficiency, decreased hepatic flow from compromised cardiac output, hyponatremia, and those taking medications that undergo CYP2D6 metabolism.
Propafenone
In addition to blocking fast sodium channels, propafenone is a weak beta-adrenergic antagonist and calcium channel blocker. See the following:
-
Indications: Atrial fibrillation and life-threatening ventricular dysrhythmias
-
Dosages: 150-300 mg every 8 hours, not to exceed 1200 mg/day; dose increases should be made at intervals of 3-4 days; sustained-release dosing is 225-425 twice daily; single dose of 600 mg may be used for the acute pharmacologic cardioversion of paroxysmal atrial fibrillation (unlabeled use)
-
Metabolism: Hepatic metabolism by CYP2D6 (predominantly), CYP3A4, and CYP1A2; genetic polymorphisms in CYP2D6 result in variable metabolism rates
-
Therapeutic concentrations: 200-500 ng/mL
-
Drug interactions: Increases serum concentrations of warfarin, digoxin, propranolol, and metoprolol; drugs that inhibit CYP2D6 or CYP3A4 can increase serum levels of propafenone
Patients at risk for propafenone toxicity include those with a polymorphism of CYP2D6 that slows metabolism, patients with hepatic dysfunction, and those taking drugs that interfere with CYP2D6 metabolism. Similarly to flecanaide, patients with structural heart disease and/or those being treated for ventricular rather than supraventricular arrhythmias are at higher risk for concerning severe cardiovascular complications such as arrtyhmia and cardiac arrest.
Class III antidysrhythmics
Amiodarone
Amiodarone blocks fast sodium channels, beta-receptors, L-type calcium channels, and delayed rectifier potassium channels. It prolongs the effective refractory periods of all cardiac tissue. Additionally, amiodarone inhibits the conversion of thyroxine to triiodothyronine. See the following:
-
Indications: Supraventricular and life-threatening ventricular dysrhythmias; atrial fibrillation prophylaxis following open heart surgery (unlabeled use)
-
Dosages: Intravenous, 150 mg over 10 minutes followed by 1 mg/min for 6 hours and 0.5 mg/min for the remaining time; oral, start with 800-1200 mg/day for the first 3 weeks (given once or twice daily) and reduce to 400 mg/day for several weeks to a maintenance dose of 300 mg or less per day
-
Metabolism: Minimal first-pass effect, excreted largely in bile; metabolized by CYP3A4 to desethylamiodarone, an active metabolite with an antidysrhythmic effect; this metabolite undergoes hepatic metabolism; onset of action after oral administration is delayed by several days
-
Therapeutic concentrations: 1-2.5 mg/mL
-
Drug interactions: concentrations are increased by digoxin, diltiazem, quinidine, procainamide, oral anticoagulants, and phenytoin; international normalized ration should be closely monitored in patients taking amiodarone and warfarin; amiodarone inhibits cytochrome enzymes and transport proteins (p-glycoprotein); QTc-prolonging drugs that undergo metabolism to these proteins can add to prolongation of the QTc interval; may enhance bradycardia in patients on beta-blockers; may also increase serum concentration of dabigatran etexilate
Amiodarone is well known to cause thyroid, liver, and pulmonary toxicity. It also has adverse CNS and skin side effects.
Dronedarone
Dronedarone is a noniodinated derivative of amiodarone, and like amiodarone it inhibits sodium channels, potassium channels, L-type calcium channels, and beta-receptors. Dronedarone also inhibits alpha1 receptors. Dronedarone is thought to cause less lung, liver, and thyroid toxicity than amiodarone. Use of this drug is contraindicated in any patient with an ejection fraction of less than 35% or class IV heart failure. See the following:
-
Indications: Atrial and ventricular dysrhythmias; maintenance of sinus rhythm in patients with atrial flutter or fibrillation and no significant cardiovascular disease; atrial fibrillation in patients with hypertrophic cardiomyopathy (unlabeled use)
-
Dosages: 400 mg twice daily
-
Metabolism: Hepatic metabolism by CYP3A4 to active and inactive metabolites
-
Therapeutic concentrations: 84-167 ng/mL
-
Drug interactions: Digoxin, beta-blockers, calcium channel blockers, any agent that prolongs QT; CYP3A4 inhibitors (azoles, cyclosporine, clarithromycin, ritonavir) may increase dronedarone toxicity; may increase international normalized ratio in patients taking warfarin; can significantly increase statin concentrations
Hepatic dysfunction increases the risk of dronedarone toxicity. Higher mortality has been shown when dronedarone is given to patients with New York Heart Association class III or IV heart failure. Cases of interstial pneumonitis or bronchiolitis obliterans with organizing pneumonia (BOOP) have been reported in dronedarone users. [10]
Despite a small increase in serum creatinine levels due to inhibition of tubular secretion, dronedarone does not impact the glomerular filtration rate and overall renal function, although it may affect the renal clearance of other medications and should be monitored in patients with preexisting renal dysfunction or on nephrotoxic medications. Furthermore renal failure may occur in setting of worsening heart failure due to dronedarone. [11]
Sotalol
Sotalol is a nonselective beta-adrenergic antagonist that prolongs the action potential and effective refractory period by blocking potassium channels. See the following:
-
Indications: Ventricular dysrhythmias, atrial fibrillation, atrioventricular (AV) nodal reentrant tachycardia, AV tachycardia
-
Dosages: IV, 75-150 mg infused over 5 hours, twice daily (not to exceed 300 mg twice daily); oral, 80-160 mg twice daily, dose may be increased gradually to 240-320 mg/day; dose adjustment should be made based on degree of renal impairment
-
Metabolism: 90-100% absorption with 100% bioavailability and no metabolism; renally excreted as an unchanged drug; baseline creatinine clearance should be measured before initiating therapy and dosage should be adjusted based on degree of renal insufficiency
-
Therapeutic concentrations: 1-4 mg/mL
-
Drug interactions: Avoid use with other QTc-prolonging drugs; bradycardia or AV block may occur if patients are concurrently taking calcium channel blockers. Hypotension may occur with concomitant use with adrenergic antagonists. On the other hand, significant hypertension has been reported with concomitant clonidine usage. Antacids containing magnesium or aluminum salts may reduce sotalol bioavailability. [12, 13]
Patients at risk for toxicity are those with renal dysfunction, with concomitant use of QTc-prolonging drugs, and women. [14]
Ibutilide
Ibutilide blocks the delayed rectifier potassium channel, prolonging repolarization. It also activates the slow inward sodium current. Ibutilide increases the refractory period of the accessory pathway, the His-Purkinje system, and the AV node. See the following:
-
Indications: Acute termination of atrial fibrillation or flutter in patients with normal heart function/structure, as well as posteroperative cardioversion; used as pretreatment in electrical cardioversion as well as in atrial fibrillation in Wolff-Parkinson-White syndrome
-
Dosages: 1 mg IV over 10 minutes; a second 1-mg dose may be given after the first dose is finished if dysrhythmia persists
-
Metabolism: Hepatic
-
Drug interactions: Avoid other QTc-prolonging drugs
The primary concern of ibutilide toxicity is its QT prolongation and increased risk for torsade de pointes. [15]
Dofetilide
Dofetilide prolongs the refractory period by blocking the delayed rectifier current. This drug effect is stronger in atrial than in ventricular tissue. See the following:
-
Indications: Conversion of atrial fibrillation or flutter to sinus rhythm; suppression of recurrent atrial fibrillation
-
Dosages: Oral doses of 0.125-0.5 mg twice daily; reduce dose in patients with reduced renal function
-
Metabolism: 50% excreted unchanged in urine; remainder undergoes hepatic metabolism (CYP3A4)
-
Drug interactions: Interactions with cimetidine, verapamil, ketoconazole, hydrochlorothiazide, prochlorperazine, megestrel, and trimethoprim have been documented
Patients at risk for toxicity are those with renal impairment, congenital long QT syndrome, electrolyte derangements (ie, hypocalcemia, hypomagnesemia, hypokalemia), and concurrent therapy with other QTc-prolonging drugs and drugs that inhibit the renal cation transport system. [16]
Class V antidysrhythmics
Adenosine
Adenosine is an extracellular signaling molecule that induces a short-duration heart block when used intravenously. Adenosine increases potassium conductance and shortens the atrial action potential duration and hyperpolarizes the myocyte membrane potential. Adenosine slows conduction in the AV node. See the following:
-
Indications: Supraventricular tachycardias (SVTs), after failure of vagal maneuvers
-
Dosages: 3 mg initial dose recommended when administered through a central venous line, to a heart transplant patient, or in patients on comcomitant dipyridamole or carbamazepine; otherwise 6-mg initial dose by rapid peripheral IV push is recommended, followed by 12 mg and another 12 mg if SVT is not broken; higher doses may be needed for patients taking methylxanthines (eg, theophylline, caffeine)
-
Metabolism: Intracellularly metabolized or phosphorylated
-
Drug interactions: Effects are antagonized by methylxanthines and potentiated by dipyridamole and carbamazepine
Adenosine is contraindicated in patients with sick sinus syndrome, second- and third-degree AV block, and atrial fibrillation down an accessory pathway (Wolff-Parkinson-White syndrome).
Epidemiology
In 2020, 1185 single exposures to antiarrhythmic drugs were reported to US poison control centers. Most exposures involved adults. There were 31 exposures resulting in major toxicity and 5 deaths. [17]
Antidysrhythmic toxicity generally affects both sexes equally. However, with sotalol some studies have found that females are at higher risk for dysrhythmia (especially for torsade de pointes). [14]
Prodysrhythmic effects occur more frequently in patients with underlying heart failure.
Older patients, in general, have a higher risk for the development of dysrhythmias than younger patients. Drug-to-drug interactions are increasing, especially in elderly patients taking multiple antiarrhythmic drugs simultaneously.
-
ECG in a patient who ingested 4 of flecainide. QRS = 200 milliseconds; QTc = 585 milliseconds. Used with permission from Lippincott, Williams & Wilkins (in Martindale JL, Brown DFM. Rapid Interpretation of ECGs in Emergency Medicine: A Visual Guide. Lippincott Williams and Wilkins; 2012).
-
Schematic of the cardiac action potential. Phase 0 depicts the the influx of sodium ions. Phases 1 and 3 correspond to the sodium-channel inactivation and the repolarizing eflux of potassium ions, respectively. Phase 2 depicts the opening of voltage-sensitive calcium channels causing a plateau in voltage.




