Background
Kindler syndrome (KS) was first described in 1954 by Theresa Kindler. Kindler syndrome is a rare subtype of epidermolysis bullosa (EB), along with EB simplex, junctional EB, and dystrophic EB. Kindler syndrome is an autosomal recessive genodermatosis characterized by congenital acral skin blistering, photosensitivity, progressive poikiloderma, and diffuse cutaneous atrophy. Mucosal manifestations are common, with frequent involvement of the oral mucosa, gingiva, and gastrointestinal tract. Various complications and associated features have been identified. See the image below.
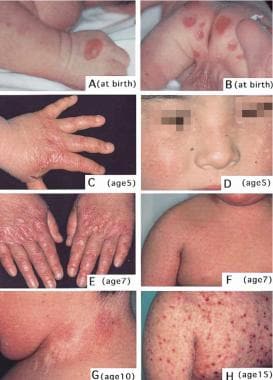 Images show the progression of lesions. A and B: At birth, acral blisters and erosions are present. C and D: At age 5 years, atrophy and reticulated erythema with dyschromic patches are noted. E and F: At age 7 years, progressive poikilodermatous changes with reticulated erythema and telangiectasia occur. G and H: At age 10 and 15 years, poikiloderma with telangiectasia and depigmentation are observed. Excoriations are due to pruritus. Reprinted from Yasukawa K, Sato-Matsumura KC, McMillan J, et al: Exclusion of COL7A1 mutation in Kindler syndrome. J Am Acad Dermatol 2002 Mar; 46(3): 447-50. Courtesy of the American Academy of Dermatology.
Images show the progression of lesions. A and B: At birth, acral blisters and erosions are present. C and D: At age 5 years, atrophy and reticulated erythema with dyschromic patches are noted. E and F: At age 7 years, progressive poikilodermatous changes with reticulated erythema and telangiectasia occur. G and H: At age 10 and 15 years, poikiloderma with telangiectasia and depigmentation are observed. Excoriations are due to pruritus. Reprinted from Yasukawa K, Sato-Matsumura KC, McMillan J, et al: Exclusion of COL7A1 mutation in Kindler syndrome. J Am Acad Dermatol 2002 Mar; 46(3): 447-50. Courtesy of the American Academy of Dermatology.
Pathophysiology
Kindler syndrome is the result of loss-of-function mutations of the FERMT1 gene (also known as KIND1).This gene has been mapped to band 20p12.3 by using linkage and homozygosity analysis in an isolated cohort of patients with Kindler syndrome. [1] FERMT1 encodes a 677–amino acid protein, kindlin-1, which binds β1 (as well as β3 and β6) integrin cytoplasmic domains. The integrin family mediates cell adhesion to the basement membrane, and absence of kindlin-1 results in poor anchoring of the basal layer of the epidermis to the underlying basement membrane. [2, 3, 4] Kindlin-1 is a human homolog of the Caenorhabditis elegans protein UNC-112, a membrane-associated structural/signaling protein that had been implicated in linking the actin cytoskeleton to the extracellular matrix (ECM). Kindler syndrome is the first genodermatosis caused by a defect in actin-ECM linkage rather than keratin-ECM linkage, underlying the pathology of other inherited skin fragility disorders (eg, other subtypes of EB). [5] Nonsense, deletion/insertion frameshift, and single-nucleotide mutations have all been described in the mutated FERMT1 gene in both introns and exons, [6, 7, 8] resulting in a truncated kindlin-1 protein. Additionally, a promoter region deletion in the FERMT1 gene has been described in a patient with Kindler syndrome. [9, 10]
Kindler syndrome keratinocytes show an up-regulation of basal level proinflammatory cytokines interleukin (IL)–1β, IL-6, and tumor necrosis factor-α (TNF-α). Ultraviolet (UV)–B irradiation to cells lacking kindlin-1 induces a greater degree of proinflammatory cytokine and p38 induction compared with control cells. UV-B–induced apoptosis has been found to be rescued by expression of kindlin-1, TNF-α blockers, a p38 inhibitor, or antioxidant plant flavonoids. [11] Oxidative stress may also result in mitochondrial dysfunction in kindlin-1–deficient keratinocytes. [12, 13] Kindlin-1 expression has also been shown to prevent premature senescence in keratinocytes, [14] as well as regulate microtuble function in mitosis. [15] Up-regulation of numerous other cytokines has been described, resulting in the activation of fibroblasts to secrete ECM proteins and differentiate into myofibroblasts. [16]
See the infographic below.
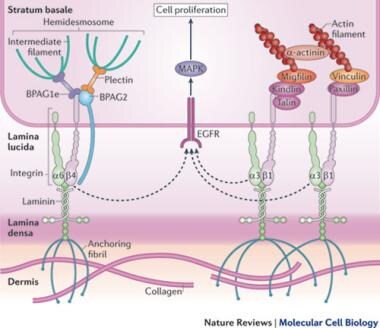 Kindlin proteins anchor intracellular actin filaments to the basement membrane via integrins. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Reprinted with permission from Nature Reviews Molecular Cell Biology (Nature Publishing Group).
Kindlin proteins anchor intracellular actin filaments to the basement membrane via integrins. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Reprinted with permission from Nature Reviews Molecular Cell Biology (Nature Publishing Group).
Epidemiology
Frequency
Since the first description in 1954, more than 250 cases of Kindler syndrome have been reported worldwide. A cluster of 26 patients with the syndrome has been identified within a tribe in the Bocas del Toro province on the northwestern Caribbean coast of Panama. [17] Owing to the rarity and autosomal recessive nature of the condition, many cases have been seen in consanguineous families, although sporadic cases are common.
Race
Persons of any race can be affected.
Sex
No sex predilection has been documented.
Age
Patients usually present with the initial skin manifestations during the first year of life. Many patients, however, may not be diagnosed until late in life.
Prognosis
Patients usually have normal intelligence and a normal life span. Blistering improves with age, but poikiloderma and atrophy are progressive. Morbidity and mortality are primarily related to secondary infections arising from cutaneous bullae and to cosmetic disfigurement.
Patient Education
Patients should be advised to avoid trauma, which helps prevent blister formation. The use of broad-spectrum photoprotection should be emphasized. Frequent consultation with a dermatologist is essential.
-
Images show the progression of lesions. A and B: At birth, acral blisters and erosions are present. C and D: At age 5 years, atrophy and reticulated erythema with dyschromic patches are noted. E and F: At age 7 years, progressive poikilodermatous changes with reticulated erythema and telangiectasia occur. G and H: At age 10 and 15 years, poikiloderma with telangiectasia and depigmentation are observed. Excoriations are due to pruritus. Reprinted from Yasukawa K, Sato-Matsumura KC, McMillan J, et al: Exclusion of COL7A1 mutation in Kindler syndrome. J Am Acad Dermatol 2002 Mar; 46(3): 447-50. Courtesy of the American Academy of Dermatology.
-
Kindlin proteins anchor intracellular actin filaments to the basement membrane via integrins. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Reprinted with permission from Nature Reviews Molecular Cell Biology (Nature Publishing Group).









