Practice Essentials
First described in 1966, the hyperimmunoglobulin E (hyper-IgE or HIE) syndrome is a rare immunodeficiency disorder that has an autosomal dominant inheritance pattern. HIE syndrome has variable expressivity and is associated with multiple abnormalities. The most common findings are recurrent skin abscesses (hence, the name Job syndrome), pneumonia with pneumatocele development, and high serum levels of IgE. Facial, dental, and skeletal features are also associated with this syndrome. A few phenotypic descriptions in the Bible from the Book of Job are said to resemble some of these patients, beyond the boils. [1, 2] See the image below.
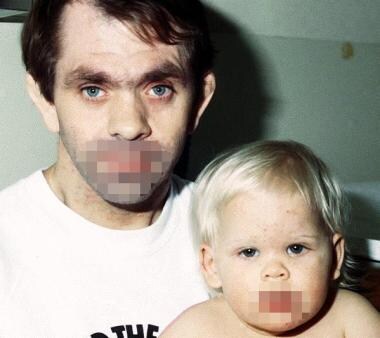 Father and daughter with autosomal dominant Job syndrome (hyperimmunoglobulin E syndrome). Note the father's distinctive facies with prominent forehead, deep-set eyes, broad nasal bridge, and wide interalar distance.
Father and daughter with autosomal dominant Job syndrome (hyperimmunoglobulin E syndrome). Note the father's distinctive facies with prominent forehead, deep-set eyes, broad nasal bridge, and wide interalar distance.
Although most cases are sporadic, multiplex families displaying autosomal dominant and autosomal recessive inheritance have been described. [3] Autosomal recessive patients tend to have severe molluscum contagiosum and other viral infections and may develop severe neurological complications. These patients also lack skeletal or dental involvement and do not develop lung cysts. Some authorities believe 2 separate syndromes exist, not one.
Type 1 HIE syndrome displays abnormalities in multiple systems, including the skeletal, dental, and immune systems, whereas type 2 HIE syndrome shows abnormalities confined to the immune system. [4, 5] Hypomorphic mutations have been found in the signal transducer and activator of transcription 3 (STAT3) gene in type 1 HIE syndrome and a null mutation in the tyrosine kinase 2 (Tyk2) gene. Cytokine responses in both types of HIE syndrome revealed severe defects leading to impaired T-helper type 17 function. Another study credited deficiency of Th17 cells in HIE syndrome to mutations in STAT3 in a majority of evaluated patients. [6] However, defective Th17 responses may be seen in classic disease without STAT3 mutations, with the extent of the defective Th17 response postulated to determine the clinical phenotype. [7]
Also see the pediatrics article Hyperimmunoglobulinemia E (Job) Syndrome.
Signs and symptoms
For systemic findings and a full discussion, please see Presentation.
Dermatologic features of Job syndrome
Nearly all patients have a history of moderate-to-severe, pruritic, eczematous skin eruptions in early life. The eruption does not have a seasonal variation and is present, to some degree, at all times.
Intermittent episodes of staphylococcal abscesses are common. These abscesses are often referred to as cold abscesses because they do not cause pain, heat, or redness.
Chronic mucocutaneous candidiasis and onychomycosis are common and usually caused by Candida species.
Dermatologic findings in Job syndrome
Moderate-to-severe, papular, pruritic eczematous lesions are typical; they may also be pustular and may become impetiginized. The areas of involvement predominately include the flexural areas, the area behind the ears, and the area around the hairline.
Cold staphylococcal abscesses that lack the typical signs of infection appear as fluctuant masses. These abscesses may be mistaken for cysts or benign tumors. They vary in size and can occur on any part of the body.
Furunculosis and cellulitis may also be present.
Chronic mucocutaneous candidiasis and onychomycosis are common.
A vesicular eruption similar to herpetic lesions may occur in newborns, with the more typical eczematous component developing over the next several months.
Disseminated molluscum contagiosum may also be evident. [8, 9]
By definition, hyper-IgE syndrome (HIE syndrome, Job syndrome) is characterized by an elevated serum IgE level. Levels vary, but the vast majority of patients have indices greater than 2000 IU/mL, and many patients have levels as high as 50,000 IU/mL. Normal values of serum IgE tend to be less than 10 IU/mL in an arithmetic distribution or less than 100 IU/mL after logarithmic conversion, although these values may vary among laboratories. Serum IgE values tend to fluctuate to some degree (most often by < 50%), and, in some patients, disease activity can significantly decrease over the years. A normal IgE level should not exclude Job syndrome in an adult.
Serum eosinophil counts are more than 2 standard deviations above the normal range of values in more than 90% of patients.
Elevated eosinophil counts can be found in secretion samples, including those obtained with abscess drainage and sputum samples in cases of bronchitis or pneumonia.
No correlation is observed between the level of serum IgE and the level of serum eosinophils, and fluctuations in these levels are not associated with infections or flares of the dermatitis.
Imaging
Pulmonary imaging (eg, radiography, CT scanning) features of Job syndrome (HIE syndrome, or hyper-IgE syndrome) typically reveal recurrent alveolar lung infections; pneumatoceles; and, rarely, pneumothorax.
Radiographs of the teeth indicate the dental development age.
Other tests
In Job syndrome (HIE syndrome, or hyper-IgE syndrome), neutrophil chemotaxis may be assessed by means of in vitro examination of their ability to move toward a chemoattractant. Such chemoattractants include endotoxin-activated serum, sodium caseinate, and formylmethionine-leucine-phenylalanine (fMet-Leu-Phe).
Although results with these tests are most often abnormal when compared with control values, chemotactic responsiveness varies, and the magnitude of the defect is less than that in other disorders (eg, Chediak-Higashi syndrome).
A polymerase chain reaction (PCR)–based high-resolution DNA-melting assay scanning selected exons of the STAT3 gene may establish a rapid molecular diagnosis in many patients. [10]
Histologic findings
In Job syndrome (HIE syndrome, or hyper-IgE syndrome), histologic examination of vesicopapules may reveal an eosinophil-rich infiltration around the hair follicles, similar to that of eosinophilic pustular folliculitis.
Management
See Treatment.
Pathophysiology
The pathophysiology of Job syndrome (HIE syndrome, or hyper-IgE syndrome) is not completely understood. [11] Patients consistently have a poor, delayed hypersensitivity response to antigens. This delayed response is associated with alterations in T-lymphocyte populations and various interleukin and cytokine abnormalities. [12] One of the earliest reports on the pathophysiology of Job syndrome described a chemotactic defect in neutrophils. [13] This defect has since been attributed to defective production of interferon-gamma, a major activator of neutrophils when stimulated by interleukin (IL)–12. The poor production of interferon-gamma in response to IL-12 results in the marked elevation of IgE levels (by means of unopposed IL-4 action). [14]
Other factors in the abnormal immunologic response are described. Patients with HIE syndrome have elevated levels of granulocyte-macrophage colony-stimulating factor, which may also explain the decreased chemotaxis and increased oxygen radical production and tissue damage. [15] Deficient suppressor T-cell numbers and activity and an imbalance in helper T cell type 1 (TH1) and helper T cell type 2 (TH2) also may play a role in an abnormal response. [16]
Although the cytokine dysregulation seems to play a role in its pathophysiology, the causative gene has not yet been identified. [17] In one study, no unique polymorphisms or mutations were found in candidate genes from the toll-like receptor pathway. [18] A significantly large number of immunoglobulin-related genes were found to be up-regulated in this syndrome. Perhaps the distinct patterns may facilitate understanding of its pathophysiology and, possibly, its diagnosis.
The hyper-IgE syndromes have multiple genetic bases. The majority of patients have dominant mutations in the signal transducer and activator of transcription 3 (STAT3) gene STAT3. [19] They do not seem to have a propensity to develop food allergies and anaphylaxis. [20] Autosomal recessive mutations in DOCK8 are linked with the autosomal recessive hyper-IgE syndrome in combination with severe atopic dermatitis, food allergies, and recurrent pneumonias. [21, 22, 23] Dominant-negative mutations in the STAT3 gene have been associated with the classic multisystem form of hyper-IgE syndrome. [24] A novel STK4 mutation has been described in patients with autoimmune cytopenias having features overlapping with those of DOCK-8 deficiency hyper-IgE syndrome. [25] A novel intronic homozygous variant in DOCK8 may produce HIE syndrome. [26]
Hyper-IgE syndrome may be associated with defective salivary activity, which accounts for the enhanced susceptibility of these patients to oral candidiasis. [27]
Etiology
Although the described defects in immune response may explain the recurrent infections and chronic dermatitis associated with Job syndrome (HIE syndrome, or hyper-IgE syndrome), the many other congenital abnormalities are not readily explained. A single-locus autosomal dominant model of inheritance with varying expressivity is described, [28] ; the greater severity of cases in younger generations of patients may suggest genetic anticipation. Findings from a multipoint analysis confirm that the proximal 4q region contains the disease locus for Job syndrome. [29]
The carrier frequency of the mutation in ZNF341 in a studied Israeli village population was found to be 1:20, which was attributed to founder mutation and consanguineous marriages. [30]
Most patients have HIE syndrome caused by dominant negative mutations in the STAT3 gene. [31] Impaired Th17 cell development may account for the cutaneous pathology.
STAT3 mosaicism may produce a milder phenotype. [32]
Epidemiology
Job syndrome (HIE syndrome, or hyper-IgE syndrome) is a rare disorder; about 250 cases have been published.
Job syndrome occurs in people of diverse ethnic backgrounds and does not seem to be more common in any specific population.
No sex predilection is reported for Job syndrome.
Job syndrome usually commences in infancy, but diagnosis is often delayed until childhood or even adulthood.
Prognosis
Few data are available on the prognosis of patients with Job syndrome (HIE syndrome, or hyper-IgE syndrome). Many Job syndrome patients who are undergoing regular monitoring and receiving appropriate treatment will live beyond the age of 50 years. Death is often due to infectious complications. The mortality rate is elevated because of systemic infections.
Significant morbidity is associated with Job syndrome. The vast majority of patients have severe cutaneous and pulmonary disease, and most patients have multiple bone fractures and scoliosis.
Patient Education
Educate patients with Job syndrome (HIE syndrome, or hyper-IgE syndrome) about the importance of recognizing the early signs of infection so that treatment can be initiated as soon as possible.
Mild local pain should be considered a sign of possible infection, and Job syndrome patients should be taught that the typical inflammatory response does not necessarily occur.
-
Father and daughter with autosomal dominant Job syndrome (hyperimmunoglobulin E syndrome). Note the father's distinctive facies with prominent forehead, deep-set eyes, broad nasal bridge, and wide interalar distance.







