Practice Essentials
Dowling-Degos disease (reticulate pigmented anomaly) is slowly progressive. It is characterized by pigmented filiform epidermal papules closely resembling an adenoid seborrheic keratosis, but similar proliferations also develop around the variably dilated pilosebaceous follicles.
Typical clinical Dowling-Degos disease (reticulate pigmented anomaly) may histopathologically be Galli-Galli disease. [1] Galli-Galli disease is a rare genodermatosis in the spectrum of reticulate hyperpigmentation, probably best regarded as an acantholytic variant of Dowling-Degos disease (reticulate pigmented anomaly). [2]
Signs and symptoms
A rare pigmentary disorder, Dowling-Degos disease (reticulate pigmented anomaly) is characterized by reticulate pigmentation of the flexures, prominent comedolike lesions and pitted scars.
Some patients report pruritus in the involved regions. Most patients in the authors' experience report increased pigmentation in the neck, the axillae, or the groin, with an onset varying from childhood to adult life. It has been described in one family as occurring together with dyschromatosis universalis hereditaria. [3]
A rare association has been described between hidradenitis suppurativa and Dowling-Degos disease (reticulate pigmented anomaly), in one case together with multiple epidermal cysts. The latter have been described alone with Dowling-Degos disease. [4] This may reflect a single underlying defect of follicular proliferation. [5]
The flexural pigmentation has its onset from childhood to adult life. It may be intense, with a brownish black color and sometimes steel blue or navy overtones. It may be generalized with truncal involvement, or be less severe, with stippled shades of brown. Rarely, the macules may be hypopigmented. [6, 7]
No verrucous or velvety papillomatosis is present, as might be seen in acanthosis nigricans. If the patches are palpable, it is because of lichenification that produces a glossy and at times somewhat wrinkled appearance. The margins may show tiny pigmented comedones.
In some cases of Dowling-Degos disease (reticulate pigmented anomaly), comedolike papules may be present on the back and/or neck. Dowling-Degos disease with follicular localization has been characterized as an uncommon variant of this evolving dermatosis. [8] Some patients with Dowling-Degos disease (reticulate pigmented anomaly) have pitted perioral scars.
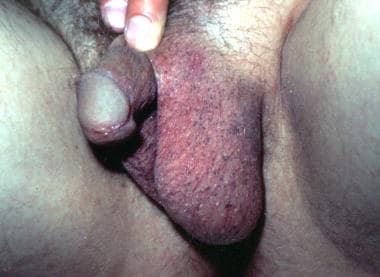
In addition, speckled macules involving the dorsum of the hands, the proximal nail folds, or the scrotum [9] may be seen. It may begin as numerous pigmented macules on the axillary and anogenital skin. [10] There may be diffuse penile pigmentation. [11] Fingernail dystrophy may be present. [12] The finding of speckled macules on the scrotum is isolated and limited to the scrotal and penile skin. [9] This pigmented eruption on the male external genitalia is possibly a cutaneous marker of underlying testicular carcinoma. In female patients, speckled macules may be found on the vulva. [13, 14, 15] Note the image below.
Diagnostics
The dermoscopic findings of Dowling-Degos disease (reticulate pigmented anomaly) are not specific, but they may be useful in selected patients to rule out melanoma of the vulva. [14]
In some patients, molecular genetic analysis can facilitate making the diagnosis. [16]
Histopathologically in Dowling-Degos disease (reticulate pigmented anomaly), pigmented rete ridge elongation with thinning of the suprapapillary epithelium, dermal melanosis, and perivascular lymphohistiocytic infiltration is present. The downward elongations are composed of regular pigmented basaloid cells.
A small study showed all pigmented cells in the basal layer were recognized by anti-PEP-1, anti-PEP-2, HMB-45, and NKI/beteb antibodies. [17] Supranuclear "caps" of brown granules were observed within most basal keratinocytes in the hyperpigmentation area. The melanocytes contained regular melanosomes in all stages of maturation in their cytoplasms. Melanosomes, mainly of stages III and IV, were evident within keratinocytes, distributed either as scattered patterns or forming caps over the nucleus. Note the image below.
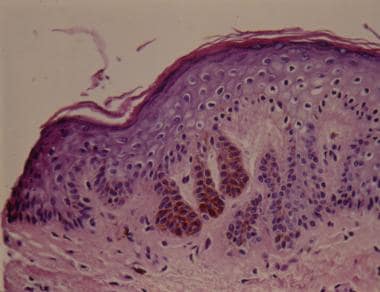 Histologic view showing elongated thin rete ridges with melanin concentrated at the tips (hematoxylin and eosin).
Histologic view showing elongated thin rete ridges with melanin concentrated at the tips (hematoxylin and eosin).
Management
No treatment is effective for Dowling-Degos disease (reticulate pigmented anomaly). [12] Topical retinoic acids, topical steroids, hydroquinone, tretinoin, and systemic retinoids have been used without success. Pruritus may respond well to H1 antihistamines. [16]
Dowling-Degos disease (reticulate pigmented anomaly) has been successfully treated with the fractional Er:YAG laser [18] and a combination of Q-switched Nd: YAG and fractional carbon dioxide laser. [19] One patient responded favorably to the use of intense pulsed light. [20]
Background
Dowling [21] first delineated this genodermatosis as a distinct entity in 1938. In 1954, Degos and Ossipowski [22] described a patient with a similar case. Few patients with reticulate pigmented anomaly, also known as Dowling-Degos disease (DDD), have been reported. [23]
Loss-of-function mutations were identified in the keratin 5 gene (KRT5) in all affected family members and in 6 unrelated patients with Dowling-Degos disease (reticulate pigmented anomaly). Another study found the same KRT5 mutation in patients with reticulate pigmented anomaly and its acantholytic variant, Galli-Galli disease. [24] This variant has a genotype/phenotype correlation with mutations in the keratin 5 (KRT5) gene. [25] In analyzing 5 patients lacking a KRT5 mutation, mutations in POGLUT1, encoding protein O-glucosyltransferase 1, were identified. [26] Additional POFUT1 mutations in Dowling-Degos disease have been documented. [27, 28, 29, 30] A novel mutation in the POFUT1 gene associated with Dowling-Degos disease and hidradenitis suppurativa has been described. [31] Scrotal Dowling-Degos disease was identified due to a novel frameshift variant in a gamma-secretase subunit presenile enhancer gene, a heterozygous PSENEN frameshift variant. [32]
Pathophysiology
Dowling-Degos disease (reticulate pigmented anomaly) is often familial and appears to be inherited in an autosomal dominant manner. [33, 34] A gene locus believed responsible in one Chinese patient was mapped to 17p13.3. [35] A genome-wide linkage analysis of 2 German families mapped this disease to 12q. [36] This region includes the keratin gene cluster, which was screened for mutations.
Loss-of-function mutations were identified in the keratin 5 gene (KRT5) in all affected family members and in 6 unrelated patients with Dowling-Degos disease (reticulate pigmented anomaly). Another study found the same KRT5 mutation in patients with reticulate pigmented anomaly and its acantholytic variant, Galli-Galli disease. [24] This variant has a genotype/phenotype correlation with mutations in the keratin 5 (KRT5) gene. [25] In analyzing 5 patients lacking a KRT5 mutation, mutations in POGLUT1, encoding protein O-glucosyltransferase 1, were identified. [26] A patient was described with POFUT1 exon deletion. [37] Additional POFUT1 mutations in Dowling-Degos disease have been documented. [27, 28]
A heterozygous frameshift mutation in the V1 domain of keratin 5 was identified in a family with Dowling-Degos disease (reticulate pigmented anomaly). [38] This study confirmed that haploinsufficiency for K5 causes Dowling-Degos disease (reticulate pigmented anomaly) and points to a prominent role for the keratin intermediate filament cytoskeleton within basal keratinocytes in epidermal pigment biology.
Etiology
See Pathophysiology. Dowling-Degos disease (reticulate pigmented anomaly) may coexist with hidradenitis suppurativa. [31] The follicular occlusion inherent in Dowling-Degos disease (reticulate pigmented anomaly) may predispose to the development of hidradenitis suppurativa. [39, 40] PUVA therapy neither aggravates nor reveals Dowling-Degos disease, [41] although it was described in one patient after this treatment.
Epidemiology
Frequency
Dowling-Degos disease (reticulate pigmented anomaly) is a rare condition worldwide.
Sex
Dowling-Degos disease (reticulate pigmented anomaly) affects both sexes. Although Dowling-Degos disease (reticulate pigmented anomaly) appears to be inherited in an autosomal dominant manner, [42] a female predominance has been noted in some surveys. [12]
Age
Dowling-Degos disease (reticulate pigmented anomaly) tends to develop early in adult life, with the onset of pigmentation occurring in individuals before they are aged 24 years. A Chinese newborn with reticulate pigmented anomaly of the flexures was recently described. [43]
Prognosis
Dowling-Degos disease (reticulate pigmented anomaly) is slowly progressive but not life threatening.
Patient Education
The patient and his or her family should be educated about the common autosomal dominant nature of Dowling-Degos disease (reticulate pigmented anomaly).
-
Speckled macules on the male external genitalia.
-
Histologic view showing elongated thin rete ridges with melanin concentrated at the tips (hematoxylin and eosin).


