Background
Vogt-Koyanagi-Harada (VKH) disease is a multisystem autoimmune inflammatory disorder with ocular, auditory, skin, and neurologic involvement. VKH disease occurs more commonly in patients with a genetic predisposition to the disease, including those from Asian, Middle Eastern, Hispanic, and Native American populations. Several human leukocyte antigen (HLA) associations have been found in patients with VKH disease, including HLA-DR4, HLA-DR53, and HLA-DQ4. (See Etiology and Epidemiology.) [1, 2, 3, 4, 5, 6, 7]
Independently, Vogt, Koyanagi, and Harada described several patients during a 20-year period with bilateral uveitis, exudative retinal detachments, neurologic abnormalities, and disorders of the integument. Despite differences in their patients, the manifestations appeared to represent a spectrum of disease, and several authors suggested that the disorder should be termed Vogt-Koyanagi-Harada syndrome (see the image below). (See Presentation and Workup.) [1, 2, 8, 9, 10, 11, 12]
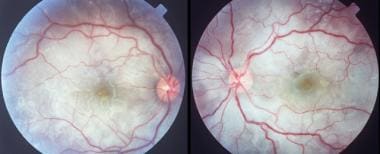 Bilateral, multifocal serous detachments in a patient with Vogt-Koyanagi-Harada disease. Disc hyperemia is evident in the right eye.
Bilateral, multifocal serous detachments in a patient with Vogt-Koyanagi-Harada disease. Disc hyperemia is evident in the right eye.
With such a wide spectrum of manifestations, typical cases of VKH disease are uncommon. To help clarify the diagnostic features of VKH disease, the International Committee on Nomenclature established revised criteria for the diagnosis of VKH disease, published in 2001. The revised criteria defined the following 3 categories of disease [11, 13] :
-
Complete VKH disease: Although involvement of all 3 systems is necessary for classification as complete disease, the neurologic and auditory manifestations often resolve before an ophthalmic examination. Patients with complete VKH disease also must have evidence of neurologic and auditory manifestations, as well as integumentary signs. However, the neurologic and auditory manifestations may resolve before an ophthalmic examination. The neurologic and auditory signs include the following: meningismus, tinnitus, integumentary signs, and cerebrospinal fluid pleocytosis.
-
Incomplete VKH disease: Patients with incomplete VKH disease have either neurologic and auditory manifestations or integumentary signs, but not both.
-
Probable VKH disease: Patients with probable VKH disease include those with isolated ocular disease.
Common to all forms of VKH disease are the following requirements, with additional criteria provided below for each form of the disease (see Presentation and Workup):
-
Patients have no prior history of ocular trauma or surgery
-
Patients have no evidence of another ocular disease based on clinical or laboratory evidence
-
Patients have bilateral ocular involvement
In March 2021, The Standardization of Uveitis Nomenclature (SUN) Working Group, published updated VKH classification criteria in an effort to standardize terminology. [14] This classification criteria allows the distinction of early and late-stage disease, as well as VKH from other panuveitides (sarcoidosis, Behcet disease, sympathetic ophthalmia, syphilis and tuberculosis). Defining features of early-stage VKH include bilateral serous retinal detachments, or, characteristic uveitis with at least 2 of the following neurologic findings: headache, tinnitus, dysacusis, meningismus, or cerebrospinal fluid pleocytosis. Defining features of late-stage disease include sunset glow fundus, or, characteristic uveitis with at least 1 of the following cutaneous findings: vitiligo, poliosis, or alopecia. Any history of penetrating ocular trauma or surgery, positive treponemal syphilis serology, or evidence of sarcoidosis needs to be excluded prior to making diagnosis of VKH.
Etiology
VKH disease is considered to be a T-cell–mediated autoimmune response directed against melanocytes, although the exact antigens involved are incompletely understood. Although the pathogenesis of VKH disease is uncertain, the wide spectrum of findings in this disorder suggests a central mechanism to account for the multisystem manifestations. Inflammation and loss of melanocytes have been described in a number of tissues, including the skin, inner ear, meninges, and uvea. These histopathologic changes suggest an infectious or autoimmune basis for the disease.
The possibility that VKH disease has an autoimmune pathogenesis is supported by the statistically significant frequency of HLA-DR4, an antigen commonly associated with other autoimmune diseases.
An autoimmune reaction seems to be directed against an antigenic component shared by uveal, dermal, and meningeal melanocytes. The exact target antigen has not been identified, but possible candidates include tyrosinase- or tyrosinase-related proteins, [15, 16, 17] an unidentified 75-kd protein obtained from cultured human melanoma cells (G-361), [15] and S-100 protein. [18] Evidence suggests that Th1 and Th17 subsets of T cells together with the cytokines interleukin (IL)-7, IL-17, and IL-23 likely are involved in the initiation and maintenance of the inflammatory process. [19, 20, 21]
VKH disease can be associated with other autoimmune disorders, such as autoimmune polyglandular syndrome, [22] hypothyroidism, Hashimoto thyroiditis, diabetes mellitus, [23, 24] Guillain-Barré syndrome, [25] and immunoglobulin A (IgA) nephropathy. [26] This syndrome also has been reported to be linked to malignant lymphoma. [27]
Immunologic analysis of cerebrospinal fluid (CSF) lymphocytes in VKH disease and studies of human uveal melanocytes show that uveal pigment can stimulate lymphocyte cultures from patients with VKH syndrome. Lymphocytes of peripheral blood and CSF from these patients may reveal in vitro cytotoxicity against allogenic melanoma cells.
Circulating antibodies against a retinal photoreceptor region have been detected in patients with this disorder.
The clinical course of VKH disease with an influenzalike illness (ILI) suggests a viral or postinfectious origin. Some studies invoke a possible role of Epstein-Barr virus reactivation [28] or cytomegalovirus infection in this disease. [29] Although a viral cause has been proposed, no virus has been isolated or cultured from patients with VKH disease. Nonetheless, Schlaegel and Morris found viruslike inclusion bodies in the subretinal fluid of a patient with the disorder. [30]
Single reports of patients developing VKH disease after cutaneous injury have been noted, [31, 32] as well as 2 cases of this condition occurring after bacillus Calmette Guérin (BCG) therapy for melanoma [33] and 1 case following surgery for metastatic malignant melanoma. [34] Case reports indicate that even an indirect trauma in melanocyte-containing tissue may induce an inflammatory response in the eye, with VKH disease following a closed head trauma. [35]
A study found that a decreased vitamin D-3 level was associated with active intraocular inflammation in patients with VKH disease. [36]
Genetics
Although almost all instances of VKH syndrome are sporadic, and familial cases are rare, some authors suggest that the condition may be inherited, probably as an autosomal recessive trait.
The strong association between VKH disease and certain racial and ethnic groups suggests that the disorder may have an immunogenetic predisposition. [37] HLA typing can be useful to identify these common genetic factors, with several HLA haplotypes apparently being more common in certain populations with VKH disease.
Among Japanese patients, HLA-DR4, HLA-DR53, and HLA-DQ4 are associated strongly with the disease. [38] In Chinese patients, HLA associations are seen with HLA-DR4, HLA-DR53, and HLA-DQ7. [39] In a mixed group of American patients, Davis and colleagues found an association with HLA-DR4 and HLA-DR53, whereas HLA-DR1 and HLA-DR4 were reported in Hispanic patients living in southern California. [40]
An association with HLA-DR4, HLA-DRw53, and HLA-DQw3 has been found in subjects of Native American ancestry, and HLA-DR4 also was found to be significantly related to VKH syndrome in White Europeans, specifically in Italian patients. [41]
Data indicate that patients with VKH disease are sensitized to melanocyte epitopes and display a peptide-specific Th1 cytokine response. Patients bearing HLA-DRB1*0405 recognize a broader melanocyte-derived peptide repertoire, so the presence of this allele increases susceptibility to the development of VKH disease. [42] In a group of French VKH disease DRB1*04-positive patients, the HLA-DRB1*0405 subtype was found in 71% of them. [43]
Epidemiology
VKH disease is uncommon, but it may be seen in Asian (primarily from eastern and southeastern Asia), Middle Eastern, Hispanic, and Native American populations. The disorder is less common in Whites and Blacks from sub-Saharan Africa. [44, 45]
In Japan, VKH disease represents 7-8% of all patients with uveitis. [46] This disorder rarely is seen in Northern European individuals. (The manifestations of VKH disease in Whites resemble those in the Japanese population, but cutaneous signs are much rarer.)
Race-related demographics
In a report from the National Eye Institute, Nussenblatt et al noted that among patients with VKH disease in the study, 50% were White, 35% were African American, and 13% were Hispanic; however, most patients had remote Native American ancestry. [47]
VKH disease is 1 of the most common forms of uveitis among darkly pigmented races. Individuals with the disorder most likely have an immunogenetic predisposition that is probably more common in certain ethnic groups with increased skin pigmentation, such as Asian, Middle Eastern, Hispanic, and Native-American populations. Moreover, VKH disease is distinctly uncommon in Africans, reaffirming that skin pigmentation alone is not a predisposing factor in the pathogenesis of the disease.
Sex- and age-related demographics
Females are more commonly affected than males; the female-to-male ratio in most large series is 2:1.
The age of onset of VKH disease has been reported to range from 3-89 years, with the maximum frequency in persons in their fourth decade of life. Although VKH disease is seen more frequently in the adult population, it has been reported in children as young as 3 years. [48, 49]
Prognosis
Long-term complications of VKH disease include reversible and irreversible vision loss. In patients with this disorder, vision loss often is due to cataracts, glaucoma, and choroidal neovascularization (with this last being a major cause of late vision loss). Patients with optic disc swelling may develop visual field defects that persist following resolution of the inflammation. [50] Final visual outcome depends on the rapidity and appropriateness of treatment. [51]
The prognosis may be improved by the use of early, high-dose corticosteroids during the acute phase of the disease, and afterward a slow tapering reduction in dosage until therapy is discontinued. In most cases, therapy should not be discontinued during the 3 months after onset of the disease, because of the high rate of recurrence during this period. Many patients require a tapering period of at least 3-6 months before the corticosteroids can be discontinued.
Ocular complications of VKH disease are more severe in children than in adults, leading to rapid deterioration in vision.
Complications
VKH disease is not associated with mortality. Complications of the disorder include the following:
-
Cataract
-
Closed-angle glaucoma - Pupillary block, forward rotation of the ciliary body
-
Subretinal fibrosis
-
Choroidal neovascularization
-
Neovascularization of the disc
-
Pigmentary changes of the fundus
-
Optic atrophy
-
Neurologic manifestations - Many of the neurologic manifestations may persist for weeks; most signs and symptoms resolve with corticosteroid therapy, although severe meningoencephalitic impairment has been reported
-
Auditory manifestations - Inner ear manifestations typically respond to corticosteroid therapy within weeks to months; hearing is restored completely in most patients with VKH disease
-
Bilateral, multifocal serous detachments in a patient with Vogt-Koyanagi-Harada disease. Disc hyperemia is evident in the right eye.
-
Fluorescein angiography of the left eye in a patient with Vogt-Koyanagi-Harada disease. Midphase is shown on the left, with multiple areas of hyperfluorescence at the level of the retinal pigment epithelium (RPE). Late phase on the same angiogram (right) reveals multiple placoid areas of hyperfluorescence at the level of the RPE and pooling of dye in the areas of serous detachment.
-
Patient with progressive dysacusis and recent onset of visual loss. Fundus photo shows a large, multifocal serous detachment of the right eye. B-scan ultrasonography reveals posterior choroidal thickening with an overlying retinal detachment.
-
Patient with progressive dysacusis and recent onset of visual loss is shown here following 6 weeks of systemic corticosteroid therapy. Diffuse depigmentation of the choroid with retinal pigment epithelium migration is seen. Residual retinal striae are present in the peripapillary region. B-scan ultrasonography shows resolution of retinal detachment and choroidal thickening.






