Background
Dermatofibrosis lenticularis (Buschke-Ollendorff syndrome) is a rare hereditary disorder of connective tissue. It is inherited as a pleiotropic autosomal dominant trait with incomplete penetrance. This condition was described for the first time in 1902 and was termed "scleroderma adultorum" by Abraham Buschke, and it has gone by several names since, including dermatofibrosis lenticularis disseminata. Buschke-Ollendorff syndrome is characterized by the presence of sclerotic bone lesions (osteopoikilosis and melorheostosis) in association with connective-tissue nevi (collagenomas and elastomas). [1, 2, 3, 4, 5, 6, 7]
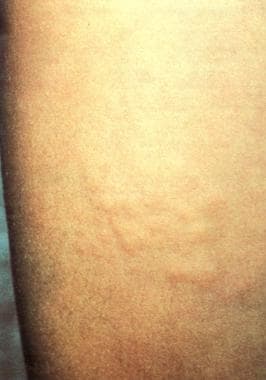
See image below depicting Buschke-Ollendorff syndrome.
Pathophysiology
The clinical findings in Buschke-Ollendorff syndrome likely result from abnormal regulation of extracellular matrix components caused by a loss-of-function mutation in the LEM domain–containing protein 3 (LEMD3) gene. LEMD3 normally functions as an inner nuclear membrane protein that antagonizes bone morphogenetic proteins and tumor growth factor-beta signaling pathways. [8] Mutations in LEMD3 lead to abnormal regulation of fibroblasts, which results in an aberrant extracellular matrix with excess elastin and thickened collagen within the dermis. [8] It has been shown that cultured fibroblasts of patients with Buschke-Ollendorff syndrome produce 2-8 times more tropoelastin than fibroblasts of healthy individuals. Elastin production is higher in both involved and uninvolved skin. [9, 10, 11] Elevated elastin mRNA levels support the notion that Buschke-Ollendorff syndrome results from abnormal regulation of extracellular matrix, leading to increased levels of elastin mRNA and increased accumulation of elastin in the dermis. Heterozygous loss-of-function mutations in the LEMD3 gene have been identified in patients with osteopoikilosis without the presence of connective-tissue nevi. [5, 12, 13, 14, 15]
Etiology
This autosomal dominant disorder is caused by a loss of function mutation in LEM domain-containing protein 3 (LEMD3) gene. [1, 4, 5]
Epidemiology
Frequency
Buschke-Ollendorff syndrome is rare syndrome, with an estimated prevalence of 1 in 20,000. [5, 16]
Race
No racial predilection is reported for Buschke-Ollendorff syndrome. [5]
Sex
The incidence is equal for males and females. [5, 16]
Age
Connective-tissue nevi and osteopoikilosis tend to first occur in the preadolescent age (average age, 7 years), but they can be found in neonates just after birth. [16]
Prognosis
In general, Buschke-Ollendorff syndrome follows a benign course. Patients are expected to have a normal lifespan. The associated lesions are generally asymptomatic and begin in childhood. They often persist throughout life and are often found as incidental findings. [17] However, proper diagnosis of this disease and careful documentation of skin and bone lesion locations can help spare the patient from any unnecessary interventions or diagnostic workup.
There have been reports associating Buschke-Ollendorff syndrome with otosclerosis (with resultant hearing impairment), stenosis of the aortae, and diabetes mellitus. [4, 8, 16, 18] Review series have also demonstrated an association of Buschke-Ollendorff syndrome with cognitive and/or developmental delay, as well as scoliosis and shortened stature. [16]
Patient Education
The patient and/or the patient's family should be educated concerning potential complications and the autosomal dominant inheritance pattern.
-
Plaque of grouped papular eruptions on the thigh.
-
Hematoxylin and eosin stain shows the histopathologic features of the skin lesions.
-
Small longitudinal lesions of increased bone density in the proximal epiphysis of the left tibial bone and in the distal epiphysis of the right tibial bone.
-
Orcein stain of elastic fibers shows the histopathologic features of the skin lesions.
-
Weigert stain of the elastic fibers shows the histopathologic features of the skin lesions.
-
Plaque of grouped papules on the abdominal coat.



