Practice Essentials
Hutchinson-Gilford progeria syndrome (HGPS) is an extremely rare hereditary disease that affects the skin, musculoskeletal system, and vasculature. HGPS is characterized by signs of premature aging most notable in the skin, cardiovascular system, and musculoskeletal systems. HGPS is caused by mutations in LMNA that result in the production of an abnormal form of lamin A termed progerin.
Background
The term progeria is derived from the Greek word geras, meaning old age. Significant morbidity and mortality result from accelerated atherosclerosis of the carotid and coronary arteries, leading to premature death during the first or second decade of life. HGPS is considered a segmental aging syndrome, as affected patients do not manifest all of the typical features of aging, such as increased incidence of cancer and neurocognitive decline.
See the image shown below depicting Hutchinson-Gilford progeria syndrome in an infant.
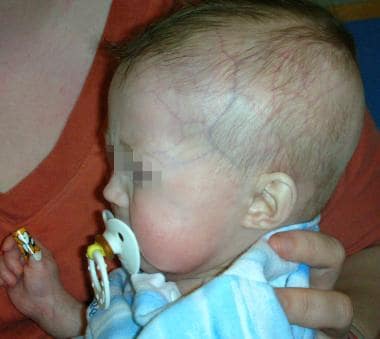 Early Hutchinson-Gilford progeria syndrome. Note the alopecia, prominent scalp veins, and frontal bossing apparent in this 12-month-old infant with Hutchinson-Gilford progeria syndrome. Midface hypoplasia and micrognathia are less apparent.
Early Hutchinson-Gilford progeria syndrome. Note the alopecia, prominent scalp veins, and frontal bossing apparent in this 12-month-old infant with Hutchinson-Gilford progeria syndrome. Midface hypoplasia and micrognathia are less apparent.
In 1886, Hutchinson [1] described the first patient with HGPS, a 6-year-old boy whose overall appearance was that of an old man. [2] In 1887, Gilford [3] described a second patient with similar clinical findings; in 1904, [4, 5] he published a series of photographs depicting the clinical manifestations of progeria at different ages. To date, approximately 100 patients with HGPS have been described in the literature.
Pathophysiology
Patients with Hutchinson-Gilford progeria syndrome (HGPS) develop clinical features of accelerated aging, including accelerated atherosclerosis of the cerebral and coronary arteries. Unlike arteriosclerosis in the general population, however, in progeria, the only lipid abnormality is decreased high-density lipoprotein cholesterol levels. Interestingly, patients with HGPS do not develop other disease processes associated with aging, such as increased tumor formation, cataract development, or senility. In this sense, HGPS is considered a segmental progeroid syndrome in that it does not recapitulate all of the characteristic phenomena of aging.
Patients with HGPS also develop loss of subcutaneous fat and muscle, skin atrophy, osteoporosis, arthritis, poor growth, and alopecia. There is evidence that patients with HGPS also manifest features of skeletal dysplasia with abnormalities in bone structural geometry and skeletal strength. [6] Extensive lipofuscin deposition, a marker for aging, is extensively distributed in patients with HGPS. Affected organs include the kidneys, brain, adrenal glands, liver, testes, and heart.
These clinical manifestations occur as the result of defects in processing and function of lamin A, an intermediate filament protein component of the nuclear membrane that regulates a diverse number of cellular functions, including nuclear morphology and integrity, DNA repair, regulation of gene expression, and telomere stability; the end result of these defects is genomic instability, decreased cell proliferation, and premature cell senescence and death. [7] The abnormal protein, progerin, represents a truncated form of the lamin A precursor prelamin A and results from mutations in LMNA. It is interesting to note that mutations in LMNA are associated not only with premature aging syndromes (HPGS, restrictive dermopathy, and atypical Werner syndrome), but also with several muscular dystrophies, lipodystrophic syndromes, and mandibuloacral dysplasia.
Marked loss of vascular smooth muscle cells within the great vessels, arteries, and arterioles associated with sclerosis and fibrosis is a consistent finding in patients with HGPS. [8] Preferential accumulation of progerin in vascular endothelial and smooth muscle cells has been observed. [9]
Clinically, children with progeria develop atherosclerosis, arteriosclerosis of small vessels, and prominent adventitial fibrosis with increasing deposition of progerin within coronary arteries. [10] The accelerated vascular stiffening and peripheral vascular occlusive disease that develop resemble the cardiovascular features of normal aging and atheroscleroisis. [11] Together with the clinical observations of accelerated and often fatal arteriosclerosis, these findings suggest that the effects of progerin on the cardiovascular system are a major contributor to the pathophysiology of HGPS.
Interestingly, spontaneous accumulation of progerin has been observed in cultured fibroblasts from normally aged individuals in combination with similar nuclear defects, further reinforcing the theory that HGPS results, at least in part, from accelerated production and accumulation of progerin. [12] It is important to note that the pathophysiology of HGPS results from the presence of progerin and a dominant-negative effect on lamin A function and not simply from the absence of normal lamin A.
Etiology
Hutchinson-Gilford progeria syndrome (HGPS) is related to aberrant processing of the nuclear envelope protein lamin A and accumulation of a farnesylated, truncated prelamin A (progerin). [13]
Autosomal dominant mutations in the LMNA gene, located on band 1q21.1-1q21.3, are responsible for most cases of HGPS. De novo mutations associated with advanced paternal age are responsible for most cases, although maternal transmission of a mutant LMNA gene from an asymptomatic mother who manifested somatic and gonadal mosaicism has also been reported. In addition, autosomal recessive transmission has also been suggested to account for the reported development of HGPS in several sets of siblings born to unaffected parents.
The LMNA genes encodes the nuclear A-type lamins, which are type V intermediate filament proteins that localize to the cell nucleus and form the nuclear lamina, a structure that supports the nuclear envelope. They are important in maintaining nuclear stability and organizing nuclear chromatin. The nuclear lamins also play a role in regulating gene expression, DNA synthesis, and DNA repair. [14]
The most common LMNA mutation and the one associated with classical HGPS involves a C-->T transition at nucleotide 1824 (G608G). Note the following:
-
This substitution results in the activation of a cryptic splice donor site in exon 11, which results in a 150-base pair deletion and a truncated lamin A protein, called progerin.
-
The abnormal progerin protein acts in a dominant-negative manner to prevent the normal assembly of nuclear lamins into the nuclear lamina.
-
After translation, the mutant preprogerin protein undergoes normal farnesylation of a CAAX tetrapeptide motif located at the carboxyterminus.
-
The farnesylated preprogerin protein is then incorporated into the nuclear membrane. However, the mutant, truncated protein lacks an important posttranslational processing signal required for cleavage of the preprogerin protein at the carboxyterminus. This cleavage is required for the release of prelamin A from the nuclear membrane, thus allowing its incorporation into the nuclear lamina. The abnormal progerin protein forms insoluble cytoplasmic aggregates.
The presence of the homozygous missense mutation G1626C (K542N) in LMNA was demonstrated in 5 siblings born to asymptomatic, consanguineous carrier parents. This study confirms that autosomal recessive inheritance of HGPS can also occur.
Somatic mosaicism for two different LMNA mutations, c.1968+2T>A and c.1968+2T>C, has been described in a child with an intermediate phenotype. [17]
A transgenic mouse model for HGPS has been created by introducing a splicing defect into intron 9 of the mouse LMNA gene. [18] Transgenic mice display many of the features of HGPS, including loss of subcutaneous fat, decreased bone density, growth failure, craniofacial deformities, skeletal abnormalities, and early death.
Using microarray analyses, 3 recent studies. [19, 20, 21] compared the gene expression profiles of cultured fibroblasts from patients with progeria with those of healthy people of various ages. In general, changes in gene activity detected in older patients correlated with changes in gene activity in progeria patients.
Of the genes expressed differentially in progeria patients, several that help control mitosis were down-regulated. Many genes that control cell division and DNA or RNA synthesis and processing were also shown to be down-regulated in progeria patients; many of these changes are also seen with normal aging. Some of these changes were postulated to lead to genetic instability and a variety of disturbances in gene function.
Changes were also seen in the expression of many genes involved in collagen remodeling and the formation of the extracellular matrix. In general, the changes favored excess extracellular matrix deposition, which may lead to the characteristic changes seen in the skin and the vasculature in progeria patients. Expression of transforming growth factor-beta, a factor that regulates tissue homeostasis and whose sustained expression is responsible for tissue fibrosis, is highly up-regulated in patients with progeria.
The expression of several transcription factors, including many involved in musculoskeletal development, were also decreased in progeria patients. Expression of MEOX/GAX, a negative regulator of cell proliferation in mesodermal tissue, is elevated almost 30-fold in patients with HGPS, suggesting a contributory role in the development of the musculoskeletal abnormalities seen in HGPS.
A characteristic finding in persons with progeria is an increase in hyaluronic acid excretion. In addition to persons with progeria, it is only detected in those with Werner syndrome, a disease characterized by a later onset of premature aging that occurs during the second decade of life.
Usually, hyaluronic acid and other glycosaminoglycan production increases during the fifth to seventh decades of life. Possibly, the increase in hyaluronic acid is a normal feature of advancing age. Fibroblasts from patients with progeria show a 3-fold increase in total glycosaminoglycan production and, in particular, hyaluronic acid production, compared with age-matched control groups. This increase results from an abnormality in degradation and is not caused by increased synthesis.
Data from embryonic development suggest that changes in the level of hyaluronic acid are extremely important for morphological development. Experiments performed in chick embryos have demonstrated a correlation between cell differentiation and hyaluronic acid degradation. Hyaluronic acid is also necessary for the morphologic development of blood vessels in chick embryos. A reduction or absence of blood vessels is noted in regions of high hyaluronic acid levels. The decreased density of vasculature, sclerodermatous changes in the skin, and the high prevalence of cardiovascular disease present in persons with progeria may be induced by increased hyaluronic acid levels. Increased hyaluronic acid levels may also promote calcification of blood vessels, thus contributing to arteriosclerosis.
In the past, studies of the link between progeria and aging (among other topics) have investigated the role of fibroblast life span.
Cells from older donors exhibit a reduced number of cell divisions in comparison to younger donor cells. The reduction of life span in cultured fibroblasts derived from patients with progeria has revealed inconsistent results. A significant reduction in fibroblast life span has been claimed in some studies but has been questioned in later investigations. A recent thorough study indicates the life span of fibroblasts in culture is independent of donor age.
Further abnormalities observed in cultured fibroblasts from patients with progeria include reduced mitotic activity, DNA synthesis, and cloning efficiency and a reduced capacity for DNA repair in cultured progeria fibroblasts after gamma irradiation. Mutant fibroblasts have been shown to demonstrate impaired DNA damage checkpoint signaling, which results in increased DNA double-strand breaks. [22]
Epidemiology
International frequency
HGPS is a rare disease with a reported prevalence of 1 in 8 million births. The true prevalence, however, has been suggested to be closer to 1 in 4 million births because many cases likely go undiagnosed or are misdiagnosed. The incidence in the Netherlands over the last century was 1:4,000,000. Approximately 100 cases of HGPS have been reported in the literature.
Race
White persons represent 97% of reported patients. The reason for this racial disparity is unknown.
Sex
HGPS has a slight male predilection; the male-to-female ratio is 1.5:1.
Age
Clinical manifestations of HGPS may not be recognized or apparent at birth, although many affected children present with sclerodermatous skin changes. Delayed recognition of the characteristic facial features along with the cutaneous and musculoskeletal manifestations may not occur until age 6-12 months or older, when the development of failure to thrive engenders a more thorough evaluation.
Prognosis
The average life expectancy for a patient with HGPS is 13 years, with an age range of 7-27 years.
Data from the largest cohort of HGPS patients indicated a mean survival of 14.6 years, with an increased mean survival of 1.6 years in patients treated with a protein farnesylation inhibitor after a median follow-up of 5.3 years from treatment initiation. [23]
Morbidity and mortality in persons with HGPS occur primarily as a result of atherosclerosis of the coronary and cerebrovascular arteries, with at least 90% of patient deaths directly related to complications of progressive atherosclerosis. Cardiovascular complications include myocardial infarction and congestive heart failure. Interstitial fibrosis, diffuse myocardial fibrosis, and calcification of the mitral and aortic valves may occur. Cerebrovascular complications occurring as a result of cerebrovascular infarction include hemiplegia, subdural hematoma, and seizures. Other causes of morbidity and mortality include marasmus, loss of mobility, and inanition.
-
Early Hutchinson-Gilford progeria syndrome. Note the alopecia, prominent scalp veins, and frontal bossing apparent in this 12-month-old infant with Hutchinson-Gilford progeria syndrome. Midface hypoplasia and micrognathia are less apparent.
-
Sclerodermatous skin changes in Hutchinson-Gilford progeria syndrome. This 12-month-old infant with Hutchinson-Gilford progeria syndrome has indurated, shiny skin with dyspigmentation.
-
Sclerodermatous skin changes in Hutchinson-Gilford progeria syndrome. This 12-month-old infant has indurated, shiny skin with dyspigmentation.
-
Enlarged joints, mild flexion contractures, and sclerodermatous skin changes are seen in this 12-month-old infant with Hutchinson-Gilford progeria syndrome.
-
Sclerodermatous skin changes in Hutchinson-Gilford progeria syndrome. This 12-month-old infant with Hutchinson-Gilford progeria syndrome has indurated, shiny skin and mild joint contractures involving the extremities and trunk.








