Practice Essentials
Encephalocraniocutaneous lipomatosis (ECCL) is a rare, congenital, neurocutaneous disorder with unilateral lipomatous cutaneous neoplasms devoid of hair and ipsilateral ophthalmologic and neurologic malformations. Haberland and Perou first described the disorder in 1970 in the clinical and necropsy findings of a 51-year-old man who has epilepsy and intellectual disability. [1] They suggested that the man had a previously unreported neurocutaneous syndrome, which they termed encephalocraniocutaneous lipomatosis. In reports, the reporting authors termed the syndrome Haberland syndrome, from the names of the original authors. Approximately 60 cases have been delineated since 1970. Early diagnosis is important to employ a needed multidisciplinary approach. [2]
Encephalocraniocutaneous lipomatosis has also become known under the term Fishman syndrome. [3]
Note the image below.
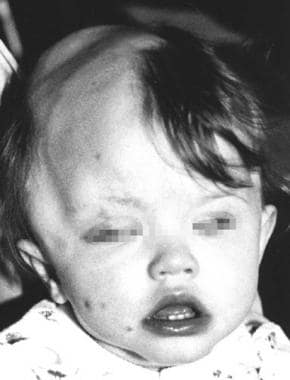 A child with Haberland syndrome. Apparent alopecia and small cutaneous soft lipomas on the face and eyelid.
A child with Haberland syndrome. Apparent alopecia and small cutaneous soft lipomas on the face and eyelid.
Etiology
All reported cases of encephalocraniocutaneous lipomatosis are sporadic. A nonhereditary, autosomal mutation that may survive only in a mosaic state may be a cause of the clinical picture of encephalocraniocutaneous lipomatosis.
Presentation
See History and Physical Examination.
Diagnostics
Laboratory test results usually are within the reference range.
Perform an electrocardiogram to exclude conduction abnormalities in children with lipomatous infiltration in the atrial myocardium.
Ophthalmologic fundus examination may reveal papilledema or optic nerve pallor. [4]
Also see Imaging Studies and Histologic Findings.
Treatment
There are no effective treatment modalities for encephalocraniocutaneous lipomatosis (ECCL).
Medical care
Therapy of cutaneous and subcutaneous lesions, especially on the face and cranium, is challenging. Zielinska-Kazmierska et al reported mandibular reconstruction after resection of a large osteoma. [5]
Antiepileptic treatment is administered in patients with clinical epileptic episodes.
Antiarrhythmic treatment may be required in patients with cardiac rhythm abnormalities.
Surgical care
Surgical treatment may be necessary in patients with extensive skin lesions. [6]
Selected patients with drug-resistant epilepsy may benefit from neurosurgical intervention, as reported by Roszkowski et al. [7]
Most spinal lipomas are asymptomatic, and surgery is usually not required. However, in some cases they may be causing neurological deficits and require surgical removal. [8] Surgical treatment may also be necessary if low-grade gliomas develop.
Consultations
Consultation with a neurosurgeon, neurologist, ophthalmologist, and cardiologist is warranted as determined by history and physical examination findings.
Long-term monitoring
Periodic neurologic and cardiologic assessment with echocardiography and electrocardiography may be indicated because of an anticipated progressive course.
Owing to reported midline low-grade gliomas in the suprasellar region in 4 encephalocraniocutaneous lipomatosis (ECCL) patients, some authors also recommend ophthalmologic and endocrinologic control in order to quickly discover any warning signs.
Pathophysiology
The pathogenesis of Haberland syndrome remains unknown. Dysgenesia of the cephalic neural crest and the anterior neural tube is a most widely accepted theory of the pathogenesis of Haberland syndrome. Haberland and Perou speculated as to whether the ectodermal malformations are caused by the same basic defect responsible for the mesenchymal malformations or whether they were secondary to the mesodermal dysgenesis. [1] No evidence exists as yet of genetic transmission or chromosomal abnormality.
Happle and Steijlen suggested that the origin of this nonhereditary genodermatosis could be a lethal autosomal mutation that survives in a mosaic state. [9]
In 2004, Cultrera et al described a female infant that showed significant overlap of encephalocraniocutaneous lipomatosis with oculocerebrocutaneous Delleman syndrome. [10] According to these authors, such overlap may support the theory of somatic mosaicism.
Legius et al reported a unique case of a child having clinical findings of Haberland syndrome and features of neurofibromatosis type 1 (NF-1), in whom a de novo mutation of the NF1 gene was detected; however, because of the relatively high incidence of NF-1 in the general population, a coincidental occurrence of both disorders in the same patient cannot be excluded. [11]
Epidemiology
Approximately 60 patients are reported in the literature. No epidemiological data on the frequency of the disorder are available. No clear racial predilection exists.
So far, the syndrome has been found in an almost equal number of girls and boys (5:6). Skin manifestations usually are evident at birth. Seizures may develop at any age and may be responsible for intellectual impairment when appearing in young children.
Prognosis
The prognosis appears to correlate with the progression of neurologic lesions, either directly or secondary to complications from drug and surgical therapies. Many patients with Haberland syndrome lead normal lives. Complications usually are caused by intracerebral malformations.
In a study by Donaire et al, functional MRI revealed transfer of memory and language functions to the nonaffected hemisphere, providing evidence that functional reorganization and restoration of cognitive function may occur in persons with encephalocraniocutaneous lipomatosis. [12]
-
A child with Haberland syndrome. Apparent alopecia and small cutaneous soft lipomas on the face and eyelid.
-
CT scan shows an enlargement of the left lateral ventricle in a child with Haberland syndrome. Note an asymmetry of the hemispheres and calcifications in the midline.
Tables
Eye |
Skin |
Central Nervous System |
Other |
Major criteria |
Major criteria |
Major criteria |
Major criteria |
Choristoma, with or without associated anomalies |
Proven nevus psiloliparis (NP) |
Intracranial lipoma |
Jaw tumor (osteoma, odontoma, or ossifying fibroma) |
Possible NP and >1 of minor criteria 2-5 |
Inraspinal lipoma |
Multiple bone cysts |
|
>2 of minor criteria 2–5 |
>2 of minor criteria |
Aortic coarctation |
|
Minor criteria |
Minor criteria |
Minor criteria |
|
Corneal and other anterior chamber anomalies |
Possible NP |
Abnormal intracranial vessels (eg, angioma, excessive vessels) |
|
Ocular or eyelid coloboma |
Patchy or streaky nonscarring alopecia (without fatty nevus) |
Arachnoid cyst or other abnormality of meninges |
|
Calcification of globe |
Subcutaneous lipoma(s) in frontotemporal region |
Complete or partial atrophy of a hemisphere |
|
Focal skin aplasia/hypoplasia on scalp |
Porencephalic cyst(s) |
||
Small nodular skin tags on eyelids or between outer and tragus canthus |
Asymmetrically dilated ventricles or hydrocephalus |
||
Calcification (not basal ganglia) |
|||
Application of the Criteria to the Diagnosis of Encephalocraniocutaneous Lipomatosis |
|||
Definite case |
|||
Three systems involved, major criteria in >2, or |
|||
Three systems involved, proven NP or possible NP + >1 of minor skin criteria 2–5 |
|||
Two systems involved with major criteria, one of which is proven NP or possible NP >1 of minor skin criteria 2-5 |
|||
Probable case |
|||
Two systems involved, major criteria in both |
|||
Two systems involved, proven or possible NP |
|||



