Practice Essentials
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive condition of chromosomal instability that is clinically characterized by microcephaly, a distinct facial appearance, short stature, immunodeficiency, radiation sensitivity, and a strong predisposition to lymphoid malignancy. [1] These patients are at high risk for developing hematological malignancies in the first two decades of life, most commonly T-cell lymphoblastic malignancies and diffuse large B cell lymphoma. [2] Mutations in the NBN (NBS1) gene located in band 8q21 are responsible for Nijmegen breakage syndrome. Nijmegen breakage syndrome is identified as entries 251260 in and 602667 in Online Mendelian Inheritance in Man (OMIM). Note the images below.
 A 6-month-old infant with Nijmegen breakage syndrome. Note microcephaly, the slightly upward-slanting palpebral fissures, and small chin.
A 6-month-old infant with Nijmegen breakage syndrome. Note microcephaly, the slightly upward-slanting palpebral fissures, and small chin.
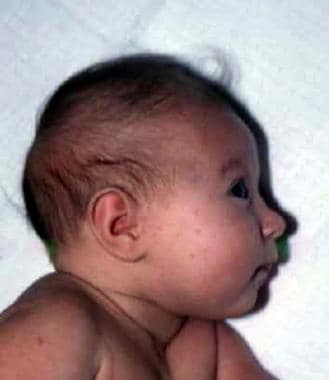 Lateral facial features with sloping forehead and receding mandible are shown in a 6-month-old infant.
Lateral facial features with sloping forehead and receding mandible are shown in a 6-month-old infant.
Patient education
Delayed speech development is observed in many children, and speech therapy is needed to correct articulation problems. Most patients with intellectual disability require educational support. They may need to attend special education classes or schools.
Signs and symptoms
Also see Presentation.
The main clinical manifestations of Nijmegen breakage syndrome include progressive microcephaly with characteristic facies, growth retardation, and impaired sexual maturation in females; recurrent infections due to a combined immune deficiency; and a strongly increased risk of developing cancer, in particular leukemia and lymphoma. Other frequently observed manifestations include skin pigmentation defects (café au lait and/or vitiligo spots) and minor limb abnormalities. [3, 4, 5, 6]
Diagnostics
Management
Also see Treatment and Guidelines.
No specific therapy is available for Nijmegen breakage syndrome.
Substitution with immunoglobulins (ie, intravenous immunoglobulin [IVIG] therapy) is indicated in patients with agammaglobulinemia (serum concentration of IgG < 2.5-3 g/L, depending on patient age) and in children with IgG2 deficiency (< 0.48 g/L in patients aged 2-5 y and < 0.72 g/L in patients >5 y). Before IVIG is started, the presence of anti-IgA antibodies must be determined in patients with IgA deficiency. In such cases, the subcutaneous administration of immunoglobulins is advocated to prevent shock, which can occur in patients with anti-IgA antibodies. A pediatric immunologist must make the decision to start substitution therapy.
Surgical care
Neurosurgical treatment with a ventriculoperitoneal shunt may be necessary for patients with hydrocephaly. Surgical repair may also be required in cases of inherited malformations (eg, anal atresia, polydactyly).
Background
In 1981, Weemaes et al [7] first delineated the syndrome in two siblings with microcephaly, short stature, skin pigmentation abnormalities, intellectual disability, immunologic defects, and a high prevalence of chromosome 7 and/or chromosome 14 rearrangements in cultured lymphocytes.
In 1985, Seemanova et al [8] described a group of patients with an apparently new genetic disorder characterized by microcephaly with normal intelligence, cellular and humoral immune defects, and a striking predisposition to lymphoreticular malignancies. These cases were subsequently studied and found to fit into the category of Nijmegen breakage syndrome.
Further investigations revealed that in vitro cells derived from patients with Nijmegen breakage syndrome display characteristic abnormalities similar to those observed in ataxia-telangiectasia (A-T), including spontaneous chromosomal instability, sensitivity to ionizing radiation (IR), and radioresistant DNA synthesis (RDS). [9, 10, 11] However, aside from immune deficiency and a predisposition for malignancies (particularly those of lymphoid origin), the clinical manifestations are distinct. Consequently, Nijmegen breakage syndrome has long been considered a variant of A-T.
In 1998, on the basis of cellular phenotypes and the results of somatic cell complementation studies suggesting genetic heterogeneity, Jaspers et al proposed the term A-T variants for diseases in this group of patients. The 2 distinct groups were designated as A-T variant 1 (V1) for Nijmegen breakage syndrome and A-T variant 2 (V2) for Berlin breakage syndrome. [12, 13]
Linkage studies allowed the exclusion of the gene responsible for Nijmegen breakage syndrome from the A-T locus on band 11q23 [14] and from the translocation breakpoints in a Polish patient. [15] The gene, NBS1 (actually named NBN), was finally mapped to band 8q21 [11, 16, 17] and cloned it in 1998, [18, 19] and mutations in this single gene were found to account for both A-T complementation groups V1 and V2. [17, 20]
Pathophysiology
Nijmegen breakage syndrome is caused by mutations in the NBN(NBS1) gene located at 8q21. The NBN gene product, nibrin, has been found to interact with at least two other proteins, hMre11 and Rad50. Nibrin plays a key role in regulating the activity of the M/R/N protein complex, which is involved in end-processing of both physiological and mutagenic DNA double-strand breaks (DSBs). DNA DSBs occur as intermediates in physiological events, such as V(D)J recombination during early B- and T-cell development and immunoglobulin class switch in mature B cells, but most frequently are generated by mutagenic agents such as IR and radiomimetic chemicals. [21, 22, 23]
DNA DSBs represent the most serious DNA damage, which, if not repaired accurately, can result in genomic instability, including chromosome rearrangements or gene mutations, and finally can lead to cancer. [24, 25] Nibrin has been shown to play a crucial role in immunoglobulin class switch recombination and maintenance of the integrity of chromosomal stability. [26, 27, 28]
Because these key regulatory processes are defective in the cells of patients with Nijmegen breakage syndrome, chromosomal aberrations accumulate and immunodeficiency and gonadal failure occur. [29, 3] However, expression study of the murine NBN gene during mouse development provides evidence that apart from sites of physiologic DSBs in the testis, thymus, and spleen, NBN expression is also evident in several tissues and organs in which rejoining of DSBs is not known to occur. [30]
Mutant murine models of Nijmegen breakage syndrome have been derived. A null mutation affecting both alleles of the homologous gene, NBN, is embryonically lethal for knockout mice. It has also been demonstrated that the common human mutation is hypomorphic and that the expression of a truncated protein is sufficient for survival. [31] Using humanized mouse models, for example with introduced the 657Δ5 mutation into the NBN gene, [32] allowed the demonstration of the pleiotropic effect of the defective protein at the cellular and organ levels.
In 2016, Seidel et al demonstrated a novel function of NBN in skin homeostasis using a mouse model with conditional postnatal inactivation of NBN in hair follicle progenitors. [33] Deficiency of NBN in hair follicle progenitors promoted a signaling DNA damage cascade and secretion of proinflammatory cytokines, leading to psoriasiform dermatitis during senescence and hair loss.
Of particular significance was the discovery of the functional link between a network of genes that play important roles in repairing DNA damage, regulating cellular proliferation and apoptosis, and maintaining telomeric function. Defects in this network, including defects in the genes encoding ATM, NBN (NBS1), BRCA1, FANCD2, BLM, TP53, CDS1/CHK2, and others, can cause cancer. [34, 35, 36]
Not all patients with the Nijmegen breakage syndrome–like phenotype and radiation sensitivity have a defect in the NBN gene. Some of these were found to have mutations in the gene encoding DNA ligase IV (LIG4), [37, 38] in the RAD50 gene, [39] , in the NHEJ1 gene, [40, 41] or the XRCC4 gene. [42, 43]
Etiology
Nijmegen breakage syndrome is a disease with an autosomal recessive pattern of inheritance.
Consanguineous matings have been reported.
The gene responsible for Nijmegen breakage syndrome, designated NBN (NBS1), is located on band 8q21.
The entire gene consists of 16 exons and spans a DNA region of more than 50 kilobases.
All disease-causing mutations identified to date have been found within exons 6-10 in the NBS1 gene and resulted in the production of a truncated protein.
More than 90% of all patients tested are homozygous for the common mutation of Slavic origin, a 5 base-pair deletion (c.657_661del5) in exon 6 of the NBN gene. [20]
The remaining patients tested to date are either heterozygous for c.657_661del5 and a second unique mutation (compound heterozygosity) or homozygous for a unique mutation. Ten unique mutations have been detected in various ethnic groups [17, 19, 20, 44, 45, 46] ; see the Table in Lab Studies.
The recent finding of the homozygous mutation c. 1089C>A in Pakistani Nijmegen breakage syndrome patients, initially diagnosed as having FA, has drawn attention to the clinical (microcephaly and congenital anomalies) and biological (increased sensitivity to both DNA cross-linking agents and IR) overlap of these 2 diseases. [47, 48]
A single case of Nijmegen breakage syndrome due to maternal isodisomy of chromosome 8 was reported. [49]
Epidemiology
Frequency
United States
The number of Nijmegen breakage syndrome patients diagnosed and molecularly confirmed within North America cannot be estimated exactly.
International
The total number of patients identified worldwide is systematically increasing, probably because physicians are becoming more aware of the disorder. The largest groups of patients were diagnosed in Poland, the Czech Republic and Slovakia, Germany, and Ukraine. Nijmegen breakage syndrome has also been reported in Italy, France, Great Britain, The Netherlands, Spain, Bosnia, Croatia, Yugoslavia, Turkey, Russia, Morocco, Argentina, Chile, and New Zealand.
The relative frequency of the common c.657_661del5 mutation in the Czech Republic, Poland, and Ukraine was studied, and it was found to be unexpectedly high in these 3 Slavic populations (a mean estimated prevalence of 1 case per 177 newborns). [50] The highest estimated frequency was reported in Sorbs, a Slavic population isolate in Northeast Saxony, Germany (1 per 34 newborns). [51]
Race-, sex-, and age-related information
Nijmegen breakage syndrome seems to occur worldwide, with an increased prevalence among persons of Eastern European and Central European descent, particularly Czech and Polish people (founder effect).
No sex predilection is recognized for Nijmegen breakage syndrome.
Microcephaly, the most striking symptom of the disease, is usually present at birth or develops soon thereafter.
Craniofacial characteristics become more obvious as patients age.
Growth is delayed from the very earliest stages of life, in comparison with age- and sex-matched controls, but improvement of the growth rate is usually observed after age 2 years.
Longitudinal studies of Polish patients indicate a decline in intellectual function with age. Most children tested during infancy and their preschool years have IQ scores indicative of normal or borderline intelligence. A shift toward a lower level of intellectual function is observed during their school-age years. This shift becomes more evident in patients older than 14 years; at this age, all tested patients had mild or moderate intellectual disability.
Progression of humoral immunodeficiency with time is observed in some children.
Most malignancies develop before patients are aged 20 years (mean age, 9 y). The youngest patient recorded to have had acute lymphoblastic leukemia was a 1-year-old girl. Cancer appears prior to the diagnosis of Nijmegen breakage syndrome in approximately 20-30% of patients.
Skin pigmentation abnormalities in the form of café au lait spots and/or vitiligo are present in more than half of Nijmegen breakage syndrome patients. Progressive vitiligo has been observed in 3 teenage patients of Polish descent.
Gray hair, which reflects progeric changes, usually appears by adolescence or early adulthood.
The longest known survival is 53 years, in an Italian woman, and 33 and 31 years in 2 men, Polish and Dutch, respectively (the latter both died from malignancy.)
Prognosis
Currently, the long-term prognosis for patients with Nijmegen breakage syndrome appears to be more positive as a result of more effective prevention, control, and treatment of infections. Premature death occurs mainly from aggressive malignancy; however, experience gained in diagnostics and management of lymphoid malignancies over the last decade has led to reduced mortality. [52] Successful bone marrow transplantation (BMT) [47] has opened a new treatment opportunity. [6]
Malignancy is the most common cause of death in patients with Nijmegen breakage syndrome. [3, 53, 4] Other known causes of death are fatal infections leading to respiratory failure, renal or liver insufficiency, [3] and bone marrow aplasia (aplastic anemia). [44]
Survival to the fifth decade has been recorded. [19]
-
A 6-month-old infant with Nijmegen breakage syndrome. Note microcephaly, the slightly upward-slanting palpebral fissures, and small chin.
-
Lateral facial features with sloping forehead and receding mandible are shown in a 6-month-old infant.
-
Typical facial features in a 9-year-old girl with Nijmegen breakage syndrome. Note the markedly upward-slanting palpebral features.
-
Lateral profile. This view shows a relatively long nose and receding mandible.
-
Cutaneous sarcoidosis in a patient with Nijmegen breakage syndrome. Note syndactyly of the second and third toes.
-
Vitiligo spots in a patient with Nijmegen breakage syndrome.
-
Progressive vitiligo in a patient with Nijmegen breakage syndrome.
-
Café au lait–like spots in a patient with Nijmegen breakage syndrome.
-
Preaxial polydactyly of the hand in a patient with Nijmegen breakage syndrome.
-
MRI in a patient with Nijmegen breakage syndrome shows large cerebrospinal fluid space that communicates with the left lateral ventricle and underdevelopment of the parietal lobes. Reprinted with permission from the Journal of Medical Genetics. Copyright 2001, BMJ Publishing Group.
-
MRI in a patient with Nijmegen breakage syndrome. Note compression of the posterior fossa and the lack of cerebellar atrophy. Reprinted with permission from the Journal of Medical Genetics. Copyright 2001, BMJ Publishing Group.
-
MRI in a patient with Nijmegen breakage syndrome. Note the small frontal lobes and the narrow frontal horns of the lateral ventricles. Reprinted with permission from the Journal of Medical Genetics. Copyright 2001, BMJ Publishing Group.
-
MRI in a patient with Nijmegen breakage syndrome. Note the partial defect of the corpus callosum. Reprinted with permission from the Journal of Medical Genetics. Copyright 2001, BMJ Publishing Group.
Tables
Clinical and Cellular Phenotype |
Nijmegen Breakage Syndrome |
|||||||
Online Mendelian Inheritance in Man (OMIM) |
251260 |
604040 |
606593 |
611291 |
616541 |
227650 |
613398 |
210900 |
Gene |
NBN |
RAD50 |
DNA LIG4 |
NHEJ1 |
XRCC4 |
FANCa |
DDX11 |
RECQL3 |
Microcephaly |
Severe, disproportionate |
Severe |
Severe |
Severe |
Severe |
In ~30% |
Severe, disproportionate |
Severe, proportionate |
Growth Deficiency |
Mild to moderate |
Severe |
Moderate to severe |
Severe |
Severe, disproportionate |
Mild to moderate |
Severe |
Severe |
Congenital Malformations |
Heart, kidney, polydactyly |
Not reported |
Polydactyly, syndactyly |
Polydactyly |
Not reported |
Heart, kidney radial bone defects (~50%) |
Heart |
Kidney, polydactyly |
Puberty and Fertility |
Primary amenorrhea (hypergonadotropic hypogonadism) |
Normal puberty |
Amenorrhea |
Not reported |
Primary amenorrhea |
Males, infertility; females, early menopause |
Normal puberty |
Males, infertility; females, early menopause |
Other Endocrinologic Problems |
Not reported |
Not reported |
Hypothyroidism, type 2 diabetes |
Not reported |
Early-onset metabolic syndrome |
Type 2 diabetes mellitus |
Not reported |
Type 2 diabetes mellitus |
Recurrent Infections |
Yes |
No |
Rare |
Yes |
No |
No |
No |
Yes |
Immunodeficiency |
Combined (B- and T-cell) |
No |
Combined (B- and T-cell) |
Combined (B- and T-cell) |
No |
No |
No |
B-cell type |
Hematologic Findings |
Myelodysplastic syndrome (incidentally) |
Not reported |
Pancytopenia, myelodysplastic syndrome |
Pancytopenia, myelodysplastic syndrome |
Thrombocytopenia, pancytopenia |
Progressive bone marrow failure, myelodysplastic syndrome |
Not reported |
Myelodysplastic syndrome |
Malignancy Type |
Predominantly lymphoid origin |
Not reported |
Predominantly lymphoid origin |
Not reported |
Tumor |
Acute myeloid leukemia, solid tumors (early onset) |
Not reported |
Lymphoid origin, acute myeloid leukemia, solid tumors |
Chromosomal Instability |
Breakages, including 7/14 rearrangements |
Breakages, 7/14 rearrangements |
Breakages, no 7/14 rearrangements |
Breakages, not specified |
Not reported |
Breakages, figures (asymmetric) |
Breakages and cohesinopathy |
Breakages, figures (symmetric), high sister chromatid exchange rate |
Sensitivity to Damaging Agents (in vitro) |
Ionizing radiation, bleomycin; mitomycin C and diepoxybutane, mild |
Ionizing radiation, bleomycin |
Ionizing radiation, bleomycin |
Ionizing radiation (variable) |
Ionizing radiation (extreme) |
Mitomycin C and diepoxybutane; ionizing radiation, mild |
Mitomycin C, camptothecin |
Ultraviolet |
Intellectual disability |
Mild to moderate |
Moderate |
Mild to moderate |
Yes, not defined |
Moderate to severe |
Moderate to severe |
Moderate to severe |
Normal to mild |
aFANC: Genetic and phenotypic heterogeneity; 19 genes known (OMIM). |
||||||||
Mutation |
Exon |
Mutation Type |
Change in Protein |
Number of Families and Origin |
Allelic Status |
c.643C>T |
6 |
Missense |
R215W |
1a Czech |
Heb |
c.657_661del5 (657del5) |
6 |
Frameshift |
Truncated protein (233 aa) |
>90% Slavic founder mutation |
Hoc (He) |
c.681delT |
6 |
Frameshift |
Truncated protein (229 aa) |
1 Russian |
He |
c.698_701del4 (698del4) |
6 |
Frameshift |
Truncated protein (236 aa) |
2 English |
Ho He |
c.742_743insGG (742insGG) |
7 |
Frameshift |
Truncated protein (251 aa) |
1 Italian |
Ho |
c.835_838del4 (835del4) |
7 |
Frameshift |
Truncated protein (279 aa) |
1 Italian |
Ho |
c.842_843insT (842insT) |
7 |
Frameshift |
Truncated protein (283 aa) |
1 Mexican |
Ho |
c.900_924del25 (900del25) |
8 |
Frameshift |
Truncated protein (305 aa) |
1 Moroccan |
Ho |
c.976C>T |
8 |
Nonsense |
Q326X |
1 Dutch |
Ho |
c.1089C>A |
9 |
Nonsense |
Y363X |
3d Pakistani |
Ho |
c.1142delC |
10 |
Frameshift |
Truncated protein (402 aa) |
2 Canadian |
He |
aMonozygotic twin-brothers (compound heterozygotes) with severe disease phenotype. [86] bHe - Heterozygous (compound with 657del5). cHo - Homozygous. dThree nuclear families in 1 large family; proband diagnosed first as having Fanconi anemia (FA). [47, 48] |
|||||




