Background
Cockayne syndrome [1] is a rare autosomal recessive (see diagram below), heterogeneous, multisystem disorder characterized by dwarfism, progressive pigmentary retinopathy, birdlike facies, and photosensitivity. The syndrome is divided into two subtypes. Cockayne syndrome I, or classic Cockayne syndrome, presents in childhood with characteristic facies and somatic features that occur late in the first decade of life. Cockayne syndrome II, or severe Cockayne syndrome, presents at birth with accelerated facial and somatic features. Individuals who are affected with Cockayne syndrome I typically have progressive neurologic degeneration with death occurring by the second or third decade of life, whereas patients with Cockayne syndrome II typically die by age 6-7 years. [2, 3]
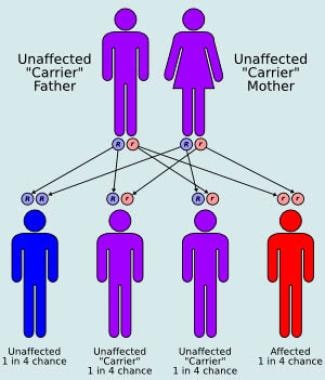 Autosomal recessive inheritance pattern.
Autosomal recessive inheritance pattern.
Also see the pediatrics article, Cockayne Syndrome.
Pathophysiology
Cockayne syndrome is an autosomal recessive disorder. A DNA repair defect is a prominent feature of Cockayne syndrome.
Cockayne syndrome, xeroderma pigmentosa, and trichothiodystrophy are 3 distinct syndromes with cellular sensitivity to ultraviolet (UV) irradiation. These syndromes arise from mutations of genes critical for nucleotide-excision repair and RNA transcription. At least 28 genes are involved in the nucleotide excision repair pathway, which is involved in protection against UV-induced DNA damage. [4, 5, 6]
Cockayne syndrome is not associated with skin cancer, despite the photosensitivity and DNA repair defect, unlike xeroderma pigmentosa. Trichothiodystrophy patients have sulfur-deficient brittle hair with a normal skin cancer risk. Progressive sensorineural deafness is an early feature of both Cockayne syndrome and xeroderma pigmentosa, but not trichothiodystrophy. Furthermore, the main neuropathology of xeroderma pigmentosa is a primary neuronal degeneration, while in Cockayne syndrome and trichothiodystrophy, myelination of the brain is reduced, suggesting that the neurological abnormalities may be caused by both developmental defects and faulty DNA repair of neuronal cells damaged by oxidative stress. [4, 5, 7, 8]
Cockayne syndrome group A or B (CSA or CSB) genes are required for transcription-coupled repair, a subpathway of nucleotide-excision repair. [9] At least 30 known CSA mutations have been characterized to date, primarily mutations in group 8 excision-repair cross-complementation gene (ERCC8) on band 5q12. [10] CSB gene defects (ERCC6) (at least 78 different mutations) result in altered expression of antiangiogenic and cell cycle genes and proteins, particularly p21, which can result in inhibition of cell cycle progression and growth. [11, 12] These may account for signs and symptoms not readily related to DNA repair deficiencies. [13, 14]
See Causes.
Etiology
Cells with a defective DNA repair mechanism are sensitive to UV light.
Decreased DNA and RNA synthesis, increased sister chromatid exchanges, and increased chromosomal breaks may occur.
In Cockayne syndrome II, the defective CS group B protein, an SNF2-family DNA-dependent ATPase, is implicated in transcription elongation, transcription coupled repair, and DNA base excision repair. [15]
Epidemiology
Frequency
Cockayne syndrome is rare worldwide.
Race
No racial predilection is reported for Cockayne syndrome.
Sex
No sexual predilection is described for Cockayne syndrome; the male-to-female ratio is equal.
Age
Cockayne syndrome I (CS-A) manifests in childhood. Cockayne syndrome II (CS-B) manifests at birth or in infancy, and it has a worse prognosis.
Prognosis
Patients with Cockayne syndrome I have progressive, unremitting, neurologic deterioration usually leading to death by the second or third decade of life. Patients with Cockayne syndrome II typically have a worse prognosis, with death occurring earlier, typically by age 6 or 7 years.
Patient Education
A genetic counselor should educate the parents of the Cockayne syndrome patient.
-
Autosomal recessive inheritance pattern.









