Practice Essentials
Homocystinuria is an inherited autosomal recessive defect in methionine metabolism that is caused by a deficiency in cystathionine synthase. [1] This defect leads to a multisystemic disorder of the connective tissue, muscles, CNS, and cardiovascular system. Homocystinuria represents a group of hereditary metabolic disorders characterized by an accumulation of homocysteine in the serum and an increased excretion of homocysteine in the urine. Note the figure below.
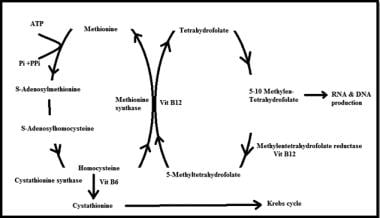 Simplified picture showing homocysteine involvement in different metabolic pathways, as well as the role of vitamins B-6, B-12, and folate as a co-factors in this pathway.
Simplified picture showing homocysteine involvement in different metabolic pathways, as well as the role of vitamins B-6, B-12, and folate as a co-factors in this pathway.
In 1960, the first case of homocystinuria was reported from Northern Ireland. The patient was initially described as having an unusual case of Marfan syndrome with renal abnormalities at age 7 years. He had recovered from acute glomerulonephritis at age 6 years and was found to be hypertensive the following year. The patient was mentally slow and thin and had fair hair, pale skin, and flushed cheeks. He had arachnodactyly, dolichostenomelia, pes cavus, a high arched palate, and bilaterally dislocated lenses. At age 10 years, the patient's urine was found to contain a large quantity of homocysteine; urinalysis results for the nitroprusside cyanide test were positive. The boy's left eye was enucleated because of a staphylococcal infection that occurred after acute pupillary-block glaucoma developed. His right lens became dislocated into the anterior chamber and had to be removed.
The patient's blood pressure readings normalized after his left kidney was removed when he was aged 13 years. Thick-walled internal arteries were noted at histologic examination. When pyridoxine supplementation was initiated at age 18 years, the patient's plasma homocysteine levels decreased below the reference range. Daily folic acid supplementation was added 1 year later because his plasma folate level was low. At age 20 years, the patient had a perforated duodenal ulcer. Chest pain occurred at age 27 years and recurred at age 34 years. The chest pain was considered to be angina and was successfully treated. At age 50 years, the patient's plasma homocysteine levels still remained low. The patient developed acute gout, which responded to indomethacin therapy.
Prognosis
The life expectancy of patients with homocystinuria is reduced. Almost one fourth of patients die as a result of thrombotic complications (eg, heart attack) before they are aged 30 years. The prognosis is favorable if patients use adequate diet alimentation.
Imaging studies
With conventional MRI, the brain abnormalities are detected in cobalamin C/D defect and include unusual basal ganglia lesions, hydrocephalus, and supratentorial white matter abnormalities. [2]
Histologic findings
DL-homocysteine inhibits the production of tyrosinase, which is the major pigment enzyme. Increased concentrations of the methionine metabolite are toxic to the nervous system. Histologic analysis of brain tissue specimens from patients with homocystinuria reveals local foci of gliosis and necrosis.
Also see Workup.
Surgical care
Surgical treatment should be considered, especially in patients with pupillary-block glaucoma or in those with recurrent lens dislocation into the anterior chamber. Other ophthalmologic or orthopedic disorders should be corrected.
Also see Medical Care and Long-Term Monitoring.
Consultations
An ophthalmologist should be consulted for the treatment of repeated lens dislocation, acute pupillary-block glaucoma, and other ophthalmologic disorders.
An orthopedist should be consulted to correct orthopedic disorders.
Diet
Patients must maintain a diet with limited amounts of protein (1 g/kg) and amino acid mixtures. The diet must be free of protein hydrolysate.
Patients in whom the disease does not respond to pyridoxine supplements must be treated with dietary reductions in methionine and with cysteine supplementation.
Pathophysiology
Homocysteine is metabolized by means of 2 pathways: remethylation and transsulfuration.
The remethylation pathway comprises 2 intersecting biochemical pathways and results in the transfer of a methyl group (CH3) to homocysteine from methylcobalamin, which receives its methyl group from S-adenosylmethionine (SAM), from 5-methyltetrahydrofolate (an active form of folic acid), or from betaine (trimethylglycine). Methionine can then be used to produce SAM, the body's universal methyl donor, which participates in several other key metabolic pathways, including the methylation of DNA and myelin.
The transsulfuration pathway of methionine/homocysteine degradation produces the amino acids cysteine and taurine. This pathway is dependent on adequate intake of vitamin B-6 and the hepatic conversion of vitamin B-6 into its active form, pyridoxal-5'-phosphate (P5P). The amino acid serine, which is a downstream metabolite generated from betaine via the homocysteine remethylation pathway is another necessary step.
Folate and vitamin B-12 are required for the remethylation of homocysteine to methionine. Findings from experimental studies have indicated that thyroid hormones affect folate metabolism. The observation that methylenetetrahydrofolate reductase is increased in hyperthyroidism and decreased in hypothyroidism may be relevant to the relationship between plasma homocysteine levels and thyroid status.
Women tend to have lower basal levels of homocysteine than do men, and neither contraceptives nor hormone replacement therapy seems to significantly alter the levels. Homocysteine concentrations are higher in postmenopausal women than in premenopausal women.
On the basis of the type of homocystinuria, the following 3 nosologic units are distinguished:
-
Homocystinuria can be caused by the deficiency of cystathionine synthase. This is the classic form. The gene for this deficiency is located on band 21q22.3. This unit includes the following forms: vitamin B-6 sensitivity (1.5% enzymatic activity), vitamin B-6 resistance (0% enzymatic activity), an intermediate variant, and a benign variant.
-
Homocystinuria can be caused by insufficient vitamin B-12 synthesis resulting from a defect in the remethylation of homocysteine to methionine; methylmalonic aciduria is present.
-
Homocystinuria can be caused by a deficiency in methylenetetrahydrofolate reductase. [3] The methionine level is in the reference range.
Urine methionine and homocysteine levels are elevated because of deficient levels of cystathionine beta-synthase. In addition to this, at least 7 causes of homocystinuria are known: (1) defect in vitamin B-12 metabolism, (2) deficiency in N -5,10-methylenetetrahydrofolate reductase, (3) selective intestinal malabsorption of vitamin B-12, (4) homocystinuria responsive to vitamin B-12 (cobalamin [cbl] type E), (5) methylcobalamin deficiency with cbl type G, (6) type 2 vitamin B-12 metabolic defect, and (7) transcobalamin II deficiency.
The basis of the disease is a defect of the gene coding for L-serine dehydratase cystathionine synthase, which converts homocysteine and serine into cystathionine. Deficient activity of this enzyme has been demonstrated in liver extracts, in brain tissue, and in cultured skin fibroblasts and lymphocytes. The deficiency leads to an accumulation of homocysteine and methionine and to its conversion into homocysteine, which is excreted in the urine (Legal test results are positive). Alternatively, methionine is reformed and detectable in appreciable amounts in the urine and serum. The accumulation of homocysteine leads to damage of the collagen and elastic fibers. The binding of homocysteine to lysine residues results in the formation of thiazine bonds.
DL-homocysteine inhibits the production of tyrosinase, which is the major pigment enzyme. Increased concentrations of the methionine metabolite are toxic to the nervous system. Histologic analysis of brain tissue specimens from patients with homocystinuria reveals local foci of gliosis and necrosis.
In 1985, Mudd et al studied hybrid cells of human fibroblasts with normal cystathionine beta-synthase activity and hamster cells without enzyme activity and found that enzyme activity was co-segregated with chromosome 21. [4] Two other enzymes involved in sulfur amino acid metabolism have been mapped: 5-methyltetrahydrofolate and L-homocysteine S-methyltransferase are mapped to chromosome 1, and cystathionase is mapped to chromosome 16. [5]
In cases of genetic deletion and partial trisomy, the levels of activity are consistent with the locus of cystathionine beta-synthase (CBS) between bands 21q22.1 and 21q21. As reported in the study of fibroblasts, 3 types of cystathionine synthetase deficiency exist; these include types with reduced activity and normal affinity for P5P and types with reduced activity and reduced affinity for the cofactor.
The human CBS gene spans more than 30 kilobases and contains 19 exons. Three different 5' untranslated regions exist in the gene.
Molecular analysis of the methionine synthase reductase (MTRR) gene in one patient reveals compound heterozygosity for a transition c.1459G>A (G487R) and a 2–base pair (bp) insertion (c.1623-1624insTA). Another patient was homozygous for a 140-bp insertion (c.903-904ins140). The insertion is caused by a T>C transition within intron 6 of the MTRR gene, which presumably leads to activation of an exon splicing enhancer. These findings support the concept that this disorder is caused by mutations in the MTRR gene.
Four different mutations were identified in patients in the United Kingdom (c.374G>A, R125Q; c.430G>A, E144K; c.833T>C, I278T; c.919G>A, G307S) and 8 mutations were identified in patients from the United States (c.341C>T, A114V; c.374G>A, R125Q; c.785C>T, T262M; c.797G>A, R266K; c.833T>C, I278T; c.919G>A, G307S; g.13217A>C (del ex 12); c.1330G>A, D444N). [6] The I278T was the predominant mutation in both populations. The spectrum of mutations observed in patients from the United Kingdom and the United States is closer to that observed in Northern Europe and bears less resemblance to that observed in Ireland.
Severe deficiency of glycine N -methyltransferase (GNMT) activity due to apparent homozygosity for a novel mutation in the gene encoding this enzyme that changes asparagine-140 to serine can be another cause of hypermethioninemia. [7]
To date, 130 pathogenic mutations have been recognized in the CBS gene. In 2004, Orendae examined 10 independent alleles in Polish patients with cystathionine beta-synthase deficiency. [8] They detected 4 already described mutations (c.1224-2A>C, c.684C>A, c.833T>C, and c.442G>A) and 2 novel mutations (c.429C>G and c.1039+1G>T). The pathogenicity of the novel mutations was demonstrated by expression in Escherichia coli. This is the first published communication on mutations leading to cystathionine beta-synthase deficiency in Poland.
Fibrillin-1 is a 350-kd calcium-binding protein that assembles to form 10- to 12-nm microfibrils in the extracellular matrix. The structure of fibrillin-1 is dominated by 2 types of disulfide-rich motifs, the calcium-binding epidermal growth factorlike and transforming growth factor beta binding proteinlike domains. Disruption of fibrillin-1 domain structure and function contributes to the pathogenic mechanisms of homocystinuria. [9]
Methylmalonic aciduria and homocystinuria, combined methylmalonic aciduria and homocystinuria (cblC) type, is the most frequent inborn error of vitamin B-12 metabolism. [10] The gene responsible for cblC, MMACHC, has been identified. Several observations on ethnic origins were noted: the c.331C>T mutation is seen in Cajun and French-Canadian patients and the c.394C>T mutation is common in the Asiatic-Indian/Pakistani/Middle Eastern populations. The recognition of phenotype-genotype correlations and the association of mutations with specific ethnicities will be useful for identification of disease-causing mutations in cblC patients, for carrier detection, and for prenatal diagnosis in families in which mutations are known. [11, 12, 13]
Homozygosity or compound heterozygosity for the c.833T>C transition (p.I278 T) in the cystathionine beta-synthase (CBS) gene represents the most common cause of pyridoxine-responsive homocystinuria in Western Eurasians. However, the frequency of the pathogenic c.833C allele, as observed in healthy newborns from several European countries (q(c.833C) approximately equals 3.3 X 10-3), is approximately 20-fold higher than expected on the basis of the observed number of symptomatic homocystinuria patients carrying this mutation (q(c.833C) approximately equals 0.18 X 10-3), implying clinical underascertainment.
The cblD gene is localized to 2q23.2, and a candidate gene, designated MMADHC (methylmalonic aciduria, cblD type, and homocystinuria), was identified in this region. Transfection of wild-type MMADHC rescued the cellular phenotype, and the functional importance of mutant alleles was shown by means of transfection with mutant constructs. The predicted MMADHC protein has sequence homology with a bacterial ATP-binding cassette transporter and contains a putative cobalamin-binding motif and a putative mitochondrial targeting sequence. Mutations in a gene designated MMADHC are responsible for the cblD defect in vitamin B-12 metabolism. Various mutations are associated with each of the 3 biochemical phenotypes of the disorder. [14]
Using data from 7038 Hordaland Homocysteine Study participants, tCys concentrations show a strong positive association with body mass index, mediated through fat mass. The link between cysteine and lipid metabolism deserves further investigation. [15]
The common polymorphism of the MTHFR gene, c.677C>T, a known risk factor for elevated plasma homocysteine levels, occurs frequently in the white persons. The sequence alteration c.677C>T combined with severe MTHFR mutations in a compound heterozygous state may lead to moderate biochemical and clinical abnormalities, exceeding those attributed to the c.677TT genotype, and might require, in addition to folate substitution, further therapy to normalize homocysteine levels. [16, 17]
Jakubowski et al showed that plasma N -Hcy-protein levels are significantly elevated in CBS - and MTHFR -deficient patients and that CBS -deficient patients have significantly elevated plasma levels of prothrombotic N -Hcy-fibrinogen. [18] These results provide a possible explanation for the increased atherothrombosis observed in CBS -deficient patients.
Homocysteine is readily oxidized in plasma to form homocystine- and homocysteine-mixed disulfides. This oxidation has been correlated with reactive oxygen species generation. Homocysteine can stimulate reactive oxygen species formation in a number of different cell types, such as splenic B lymphocytes, mesangial cells, monocytes, and vascular smooth muscle cells. The oxidative stress might participate, at least in part, in the pathophysiology of homocystinuria. Homocysteine has been found to induce neurological dysfunction via oxidative stress. The cytotoxicity of homocysteine has been reported to be mitigated by antioxidants like N -acetyl cysteine, vitamin E, or vitamin C. The cells from homocystinuria patients with defects in the remethylation pathway showed high reactive oxygen species and apoptosis levels. [19]
Chang et al have described a Taiwanese infant boy with early-onset cblC disease, heterozygous for c.609G–>A and c.567dupT, who was presymptomatic at newborn screening but later showed life-threatening manifestations. He was the first Asian and was the second case with c.567dupT mutation in the literature. Moreover, all reported cblC patients with the c.609G–>A mutation have been East Asians so far; thus, authors suggest that c.609G–>A should be included in the initial mutation screening tests for a cblC patient in East Asian populations. [20]
Epidemiology
Frequency
Homocystinuria rarely occurs. The prevalence of patients with clinically ascertained CBS deficiency is only 1 in 330,000 worldwide. The molecular epidemiological studies suggest an incidence of around 1 in 10,000 in several European populations. Expanded newborn screening in the Madrid region proved fruitful. [21]
In Ireland, the frequency is higher, specifically 1 case per 65,000 population based on newborn screening and clinically detected cases. A surprisingly high prevalence of the CBS 833T-C mutation was detected among newborns who did not carry the 844ins68 variant, which is known to neutralize the 833T-CV mutation. This finding led some authors to suggest that the incidence of homocystinuria due to homozygosity for the mutation may be at least 1 case per 20,500 live births in Denmark.
The 0.5% frequency of c.1105 C>T alleles in a predominantly Slavic population of the Czech Republic is similar to the 0.8% frequency in Norwegian newborns or 0.5% shown for 200 North American adult control subjects. Although data from other European populations are lacking, similar frequencies in unrelated Norwegians, Czechs, and North Americans suggest that this variant allele may be of ancient origin and that it may be common in populations of European descent. This high population frequency of mutant CBS alleles may have important consequences for newborn screening. The expected frequency of homocystinuria because of 6 mutations in Norway and 11 mutations in the Czech Republic are similarly high, being 1 in 6,400 and 1 in 15, 500, respectively. [22]
Hypermethioninemia was reported in Korea in 2 compound heterozygous siblings with deficient activity of methionine adenosyltransferase (MAT) in their livers (MAT I/III deficiency). Molecular genetic studies demonstrate that each patient is a compound heterozygote for 2 mutations in MAT1A, the gene that encodes the catalytic subunit that composes MAT I and MAT III. These mutations include a previously known inactivating G378S point mutation and a novel W387X truncating mutation. W387X mutant protein, expressed in E coli and purified, has about 75% of wild-type activity.
Lu et al found an inordinately high prevalence of homocystinuria in the Taiwanese-Austronesian aboriginal tribe of Orchid Island. The prevalence of homocystinuria in the Tao tribe, estimated at approximately 1 in 240 islanders, is the highest known worldwide. All patients were homozygous for the p.D47E mutation. This mutation identified in this population suggests that it interferes with the function and stability of the CBS enzyme. [23, 24]
Sex and age
The disease is more common in males than in females, and the condition is congenital.
-
Simplified picture showing homocysteine involvement in different metabolic pathways, as well as the role of vitamins B-6, B-12, and folate as a co-factors in this pathway.


