Practice Essentials
Ataxia-telangiectasia (A-T) is an autosomal recessive, complex, multisystem disorder characterized by progressive neurologic impairment, cerebellar ataxia, variable immunodeficiency with susceptibility to sinopulmonary infections, impaired organ maturation, x-ray hypersensitivity, ocular and cutaneous telangiectasia (see image below), and a predisposition to malignancy. The disease is heterogeneous, both clinically and genetically, as shown by the existence of 4 complementation groups (A, C, D, E). The responsible gene (ATM gene) has been mapped to band 11q22-23. [1]
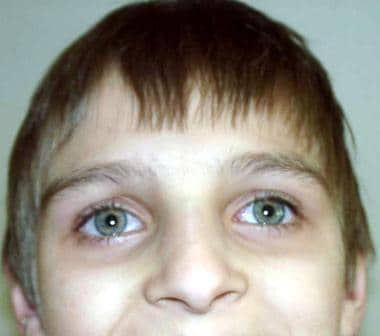
The clinical and immunological presentation of ataxia-telangiectasia may differ even within the same family, as described by Soresina et al. [2]
Syllaba and Henner first published descriptions of patients with ataxia-telangiectasia in 1926. [3] They observed progressive choreoathetosis and ocular telangiectasia in 3 members of a single family. A gap of some 15 years occurred before the next report in 1941 by Louis-Bar, who described progressive cerebellar ataxia and cutaneous telangiectasia in a Belgian child. [4] The syndrome subsequently received the name of Louis-Bar. Ataxia-telangiectasia was not described as a distinct clinical entity for another 16 years until Boder and Sedgwick [5] in 1957 and Biemond [6] in 1957, with the aid of autopsies, reported organ developmental abnormalities; neurologic manifestations; and a third major feature of the disease, recurrent sinopulmonary infection.
Ataxia-telangiectasia can best be classified, according to its major clinical and pathologic features, as a predominantly cerebellar form of spinocerebellar degeneration, which is transmitted as an autosomal recessive trait and evolves ultimately to include motor neuron disease, with spinal muscular atrophy and peripheral neuropathy.
Ataxia-telangiectasia can also be classified among the neurocutaneous syndromes, although not among the phakomatoses as originally proposed, because the vascular and cutaneous lesions of ataxia-telangiectasia are not congenital nevi but develop in the course of the disease as a progeric manifestation. Ataxia-telangiectasia should be considered among the immunodeficiency diseases, cancer-prone genetic disorders, chromosomal instability syndromes, disorders with abnormal radiosensitivity, syndromes with possible DNA-repair/processing defects, and (as is now evident) the progeroid syndromes. These patients are at high risk for developing hematological malignancies in the first two decades of life, most commonly T-cell lymphoblastic malignancies and diffuse large B cell lymphoma. [7]
Elevated immunoglobulin M (IgM) occurs in only 60% of patients, challenging this finding as a probable diagnosis criterion. [8]
Also see Ataxia-Telangiectasia in Ophthalmology.
Prognosis
Most patients are wheelchair dependent by age 10-15 years, but mild forms are not rare. Unfortunately, most patients with A-T have life limited to teenage years or early adulthood. [9]
Also see Prognosis.
Etiology
The ataxia-telangiectasia gene has been localized to band 11q22-23. The gene, called ATM (ataxia-telangiectasia mutated), is a member of a family of phosphatidylinositol-3-kinase–related genes involved in cell cycle control, intracellular protein transport, and DNA damage response. Little correlation exists between the level of ATM protein and the type of underlying mutation, clinical phenotype, or radiophenotype. The discovery of this gene has also led to phenotypic spectrum expansion. [10]
Also see Laboratory Studies.
Patient education
Good patient and parental education should include tactful genetic counseling and an explanation of the multisystem nature of the disease.
Pay special attention to the susceptibility of adult members of families with ataxia-telangiectasia to malignant neoplasms and to the importance of regular examinations for early cancer detection.
Complications
Early death is frequently due to pulmonary disease, but malignancies are also a common cause. The incidence of malignancy is 60-300 times higher than in healthy persons, and, on autopsy report, 49% of cases had malignant tumors. The most common tumors are lymphoreticular malignancies, especially non-Hodgkin lymphomas, but other kinds of tumors also occur. Malignancies are also more common in obligate heterozygotes than in the general population.
Treatment
Although no specific treatment is available, several features of ataxia-telangiectasia are accessible to active therapy. This applies especially to infections. Correction of ATM mutations may hold promise for the future. [11]
Also see Medical Care.
Activity
Daily participation (to tolerance) in a structured physical fitness program, which may include swimming, use of a special bicycle, and graduated weight lifting, is useful in maintaining good muscular strength and preventing limb contractures and, thus, may postpone confinement to a wheelchair.
Long-term monitoring
Patients with ataxia-telangiectasia should undergo regular examinations for early cancer detection.
Consultations
Provide genetic counseling to all patients with ataxia-telangiectasia and their family members.
Consult a neurologist, a cardiologist, and an endocrinologist as determined by the patient's history and physical examination.
Rehabilitation and adequate educative support are always necessary. Physical therapy is useful in maintaining good muscular strength, preventing limb contractures, and learning techniques of falling to avoid injury. Occupational therapy helps to develop functional adaptions in the activities of daily living. Speech therapy may be useful in improving articulation and in increasing voice volume.
Pathophysiology
The ATM gene encodes the protein kinase ATM, which is the key regulator of cellular response to double-strand breaks (DSB) in DNA. Therefore, ataxia-telangiectasia symptoms include all the possible consequences of the perturbations in DNA damage response (DDR). [12, 13, 14]
One basic defect associated with the malady is the abnormal sensitivity of ataxia-telangiectasia cells to x-rays and certain radiomimetic chemicals but not to ultraviolet irradiation, which leads to chromosome and chromatid breaks. Breakpoints are randomly distributed, but nonrandom chromosome rearrangements selectively affect chromosomes 7 and 14 at sites that are concerned with T-cell receptors and heavy-chain immunoglobulin coding and with the development of hematologic malignancies. Such disturbances could account for the frequency of infections and neoplasias.
As has been shown by Guerra-Maranhao et al, ataxia-telangiectasia patients are at high risk of having impaired responses to infection with pneumococci, which may be one of the causes of recurrent sinopulmonary infections in these patients. [15] The authors analyzed the production of antibodies to polysaccharide antigens in patients with ataxia-telangiectasia and found that the levels of immunoglobulin G (IgG) antibodies to serotypes 1, 3, 5, 6B, 9V, and 14 of Streptococcus pneumoniae before and after immunization with 23-valent polysaccharide vaccine were significantly lower than in a healthy population.
ATM gene targets include well-known tumor suppressor genes such as TP53 and BRCA1, both of which play an important role in the predisposition to breast cancer. Studies of ataxia-telangiectasia families have consistently reported an increased risk of breast cancer in women with 1 mutated ATM gene, [16] but, to date, an increased frequency of ATM mutations has not been found in women with breast cancer. [17]
ATM mutations are poor prognostic factor in patients with lung cancer. [18]
The mechanisms responsible for neurologic disease, thymus aplasia, telangiectasias, growth retardation, and impaired organ mutation have not been elucidated, but most likely, they are linked to accelerated telomere loss. [19, 20] ATM has been shown to be pivotal for neurodevelopment, especially for stem cell differentiation, as well as for elimination of damaged postmitotic cells. [21] Frappart and McKinnon showed that the ATM protein has a proapoptotic function in the developing mouse CNS, acting in cooperation with another key proapoptotic factor—Bax protein. [22] ATM-dependent apoptosis occurred only in postmitotic populations of neurons after irradiation.
These results suggest that ATM may serve to eliminate neurons with excessive DNA damage during CNS development. A general disturbance in tissue differentiation accounts for the almost constant elevation of alpha-fetoprotein (AFP), a fetal serum protein of hepatic origin that indicates dedifferentiation of liver cells.
Research suggests that ataxia-telangiectasia may be associated with dysregulation of the immunoglobulin gene superfamily, which includes genes for T-cell receptors. The normal switch from the production of immunoglobulin M (IgM) to IgG, immunoglobulin A (IgA), and immunoglobulin E (IgE) is defective, and the same may apply to the switch from immature T cells that express the gamma/delta rather than the alpha/beta receptors. Conceivably, an absence or a mutation of a single protein coded for by chromosome 11 could explain the immunologic and perhaps even the neurologic features of the disease. The ATM protein apparently controls the cell cycle and plays a major role in the protection of the genome.
The ATM gene product has been shown to be required for cell survival and genomic stability maintenance following exposure to low labile iron concentrations. Because iron chelation agents increase ataxia-telangiectasia cell genomic stability and viability and activate ATM-dependent cellular events in normal cells, Edwin Shackelford et al suggested that pharmacological manipulation of ATM activity via iron chelation might have clinical efficacy in Parkinson disease treatment. [23] Targeted next-generation sequencing is a rapid cost-effective method that identified five disease-causing variants in 3 Chinese probands in 1 study. [24]
Epidemiology
Frequency
Ataxia-telangiectasia is reported in all regions of the world. The probable incidence of ataxia-telangiectasia is about 1 case in 100,000 births. [25] The frequency of ataxia-telangiectasia mutant allele heterozygosity was reported to be 1.4-2% of the general population. [25, 26] The incidence of ataxia-telangiectasia was significantly higher in the highly consanguineous Bedouin population than in the relatively nonconsanguineous Jewish population of southern Israel. [27]
Race-, sex-, and age-related information
Ataxia-telangiectasia is reported in all races, although the mortality ratios differ between the ethnic groups.
Ataxia-telangiectasia occurs equally among males and females.
No characteristic features are detectable during very early childhood.
Ataxia is usually a first diagnostic hallmark, having its onset in the first years of life. Beyond the age of 5 years, the progression of the ataxia becomes increasingly apparent and the child requires a wheelchair by age 10 or 11 years. Trimis et al reported a 6-year-old girl without any neurological symptoms. [28]
Oculocutaneous telangiectasia, the second diagnostic hallmark of ataxia-telangiectasia, usually has a later onset than the ataxia, typically at age 3-6 years.
The progression of the disease is apparent in subsequent years.
Prognosis
The neurologic features of ataxia-telangiectasia are relentlessly progressive. In addition to the classic early features, older patients tend to develop other signs of spinocerebellar degeneration (eg, posterior cord involvement with loss of the deep tendon reflexes, spinal muscular atrophy). Most patients are wheelchair dependent by age 10-15 years, but mild forms are not rare.
Gene therapy holds promise for the future. [29]
A 20-month-old girl with T-cell acute lymphoblastic leukemia and ataxia-telangiectasia was apparently cured after only 7 weeks of antileukemic therapy, as she was reported in remission for 8 years. [30]
Death typically occurs in early or middle adolescence, usually from bronchopulmonary infection, less frequently from malignancy, or from a combination of both. The median age at death is reported to be approximately 20 years. [26] To date, the longest reported survival is 34 years. [31] In a retrospective study in the United States, mortality from all causes in ataxia-telangiectasia was 50- and 147-fold higher for white and black patients with ataxia-telangiectasia, respectively, than expected based on overall US mortality rates. [32]
Boder reviewed 58 complete autopsy cases; 27 (46%) deaths were caused by pulmonary infection alone, 12 (21%) by malignancy alone, 16 (28%) by a combination of both, and 3 (5%) by other reasons. [33]
The lifetime risk of cancer among patients with ataxia-telangiectasia has been estimated to be 10-38%, [34, 35, 36] which is about 100-fold more than the population rate [32] ; however, in the absence of chronic bronchopulmonary disease and lymphoreticular malignancy, ataxia-telangiectasia is consistent with survival into the fifth or sixth decade.
Ataxia-telangiectasia heterozygotes present an excess risk of death (they die 7-8 y earlier than the normal population), mostly from ischemic heart disease (ataxia-telangiectasia carriers die 11 y younger than noncarriers) or cancer (ataxia-telangiectasia carriers die 4 y younger than noncarriers). They have an enhanced risk of recurrent infections. [26, 37]
-
Face of a boy with ataxia-telangiectasia. Apparent ocular telangiectasia.
-
Close-up view of advanced telangiectasia of the bulbar conjunctiva.
-
Chest MRI showing a hyperintense lesion in the right mediastinum corresponding to lymphoma.




