Coagulation Disorders
Antiphospholipid syndrome
Antiphospholipid syndrome (APS) is an acquired, multisystemic disorder characterized by recurrent thromboses in the arterial system, venous system, or both. Antiphospholipid syndrome is classified into 2 groups: primary and secondary. [1, 2]
Secondary antiphospholipid syndrome is often associated with systemic lupus erythematous (SLE) and infrequently with other diseases, such as lymphoproliferative disorders, autoimmune diseases, infections (eg, syphilis, HIV, hepatitis C), and drugs (eg, procainamide, quinidine, hydralazine, phenytoin, chlorpromazine). Serologic markers for antiphospholipid syndrome are antiphospholipid antibodies (beta2-GPI or anticardiolipins) or lupus anticoagulant.
The APS diagnosis should meet the revised Sapporo criteria (also called the Sydney criteria) for definite APS. The criteria, developed by consensus among international clinical and laboratory experts, were originally referred to in 1999 as the Sapporo criteria [3] and then modified at a meeting in Sydney in 2006. [4]
The primary diagnostic criteria include arterial thrombosis, venous thrombosis, recurrent fetal loss, and thrombocytopenia. One of the listed primary criteria is required for diagnosis, combined with a sustained elevated titer of immunoglobulin G (IgG) anticardiolipin or lupus anticoagulant, which can be detected based on the prolonged activated partial thromboplastin time (aPTT), Kaolin clotting time, or dilute Russell Viper venom time.
In a large retrospective study from the Mayo Clinic, 41% of patients with lupus anticoagulant had skin lesions as the first sign of antiphospholipid syndrome. [5] Cutaneous manifestations include livedo reticularis, necrotizing vasculitis, livedo vasculitis, thrombophlebitis, cutaneous ulceration and necrosis, erythematous macules, purpura, ecchymoses, painful skin nodules, and subungual splinter hemorrhages.
Livedo reticularis is a presenting sign in up to 40% of patients with the diagnosis of SLE. [6] Skin changes defined as livedo reticularis are violaceous, red or blue, reticular, or mottled pattern of the skin of the arms, legs, and the trunk. They are not reversible with rewarming. [4]
Noninflammatory vascular thrombosis is the most frequent finding in skin lesions of patients with antiphospholipid syndrome. Differential diagnoses include cryoglobulinemia, warfarin-induced necrosis, purpura fulminans, emboli to the skin, thrombocythemia, protein C deficiency, Sneddon syndrome, and skin ulcers in patients with sickle cell anemia or hemolytic anemia. In some antiphospholipid syndrome lesions, hemosiderin deposition can make differentiation from Kaposi sarcoma difficult.
The mainstays of prophylaxis and treatment of thrombosis in patients with antiphospholipid syndrome are anticoagulant and antiplatelet agents. Immunosuppression and immunotherapy (eg, corticosteroids, pulse cyclophosphamide, plasmapheresis, gamma-globulin infusions) only transiently reduce elevated antibody levels. Indications for immunotherapy are limited to systemic lupus erythematous or catastrophic vascular occlusion syndrome.
No studies are available to show the usefulness of prophylaxis in patients with antibodies in the absence of thrombosis. Some authors suggest using low-dose aspirin in patients with persistent lupus anticoagulant or high levels of IgG anticardiolipin. Some patients with antiphospholipid syndrome may have resistance to a usual dose of subcutaneous heparin. Low molecular weight heparins may prove beneficial because of improved safety and efficacy profiles (eg, decreased bleeding, risk of osteoporosis and thrombocytopenia, prolonged half-life, increased bioavailability). The protective effect of hydroxychloroquine against thrombosis has been reported in patients with systemic lupus erythematous.
Regarding treatment, a lack of consensus exists concerning the intensity and the duration of anticoagulation for patients with antiphospholipid-antibody–associated thrombosis. Warfarin is the treatment of choice for patients with antiphospholipid syndrome. Moderate-intensity anticoagulation with warfarin (to an international normalized ratio [INR] of 2.0-3.0) with or without low-dose aspirin of 75 mg/d was as effective as high-intensity warfarin (to INR 3.0-4.0) in a randomized trial. [7] High-intensity warfarin therapy should be considered only in patients receiving moderate-intensity warfarin with recurrent thrombosis.
Patients with catastrophic antiphospholipid syndrome usually receive a combination of anticoagulants, corticosteroids, intravenous immunoglobulin (IVIG), and plasma exchange; however, despite this aggressive approach, the mortality remains high.
The Antiphospholipid Syndrome Alliance for Clinical Trials and International Networking, formed in 2010, concerns large-scale, multicenter clinical trials and research in persistently antiphospholipid antibody‒positive patients. [8]
The 14th International Congress on antiphospholipid antibodies held in Rio de Janeiro in September 2013 systematically reviewed the new trends in treatment strategies, including new oral anticoagulants, hydroxychloroquine, statins, rituximab, and other B-cell–targeting agents. [9]
Warfarin-induced skin necrosis [10]
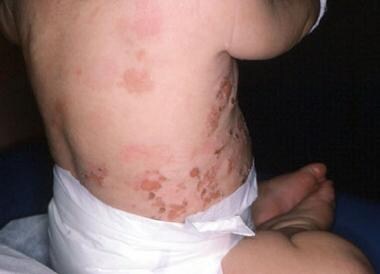
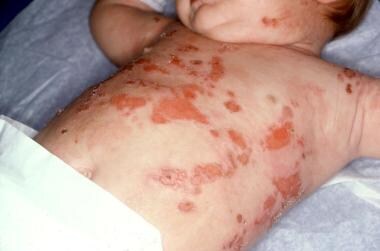
Warfarin-induced skin necrosis is an uncommon serious complication of oral anticoagulation therapy (see the image below). [11, 12, 13]
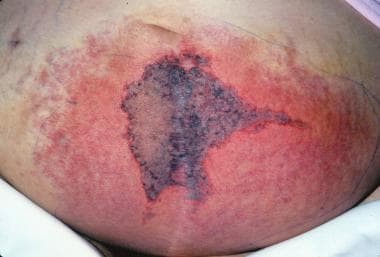
Warfarin-induced skin necrosis is estimated to occur in 0.01-0.1% of patients treated with anticoagulants. Approximately 85% of reported patients are women. The most commonly affected sites are the breasts, buttocks, thighs, and abdomen, possibly because of the abundance of small dermal blood vessels in fatty tissue. Less usual locations include the external ear skin, [14] eyelids, [15] penis, [16] or digits. [17] A classic pattern of skin changes occurs when the condition commences. The initial nonspecific symptom may be a sensation of pain or cold in the affected area, followed by the development of a well-demarcated erythematous lesion that progresses to bullae formation and full-thickness skin necrosis. Areas of erythematous flush may become edematous and have a peau d'orange effect. The eschar may eventually slough or require extensive surgical debridement.
Most cases of warfarin-induced skin necrosis occur within the first week of warfarin administration, although rare cases of late onset are reported. [18, 19] The condition has also been observed in patients who discontinue long-term warfarin therapy and then restart the drug. [20] Biopsy specimens of lesions reveal involvement of the dermis and the subcutaneous tissue characterized by fibrin deposition in the small veins and postcapillary venules, with hemorrhage and diffuse necrosis in the dermis and the subcutaneous fat. Absence of arterial thrombosis is another characteristic feature.
Differential diagnoses include necrotizing fasciitis, venous gangrene, pyoderma gangrenosum, and cholesterol embolization syndrome. Warfarin is believed to induce a relative hypercoagulable state by interfering with the natural anticoagulant pathways (eg, those involving protein C, protein S, and antithrombin). A substantial minority of cases occurs in association with a familial deficiency of protein C or protein S. An acquired deficiency of protein S secondary to the development of antiphospholipid antibodies has been implicated. There is increasing recognition of the fact that skin necrosis could be the first presentation of heparin-induced thrombocytopenia. [21, 22]
Patients receiving large, rapidly administered loading doses of warfarin are at particular risk. Identifying patients at risk (eg, patients with a previous episode, protein C or protein S deficiency, or antiphospholipid syndrome), avoiding excessive initial doses of warfarin, and administering vitamin K in the early stages of lesion development can prevent warfarin-induced skin necrosis. If lesions have already progressed, oral anticoagulation agents should be discontinued. In some patients, attempts have been made to reverse the anticoagulation with plasma, and, in patients with documented protein C deficiency, concentrates of protein C have been used. Anticoagulation with heparin should be continued until the necrotic lesions heal. Despite treatment, approximately 50% of patients ultimately require skin grafting.
Warfarin also has been implicated in the etiology of calciphylaxis in patients with end-stage renal disease. It is a poorly understood disorder associated with high morbidity and mortality (60-80%). Clinically, patients present with painful, necrotic skin ulcers. Histopathology evaluation reveals medial calcification and intimal hyperplasia of small and medium-sized arteries of dermal and subcutaneous tissues. [23] The differential diagnosis includes vasculitis, cholesterol embolization syndrome, warfarin-induced skin necrosis, nephrogenic fibrosing dermopathy, ecthyma, cryofibrinogenemia, cellulitis, necrotizing fasciitis, coagulation disorders, and adverse drug effects. Treatment options are limited. [24]
Cutaneous Manifestations of Anemia
Pallor of the skin and mucocutaneous membranes can be a sign of severe anemia of any cause. Severe anemia (hemoglobin < 7 g/dL) can be diagnosed in individuals with the loss of pink in the palmar creases on the full open palms.
Iron deficiency anemia
Iron deficiency anemia can cause changes in the appearance of the nails, tongue, and hair. In iron deficiency, nails become fragile and develop longitudinal ridges. Nail-plate alterations follow, with the development of koilonychia, a spoonlike convexity (see the images below).
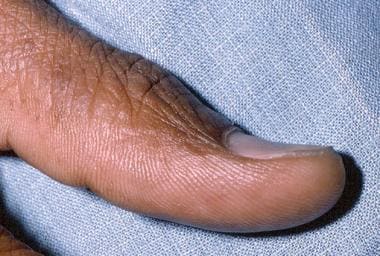
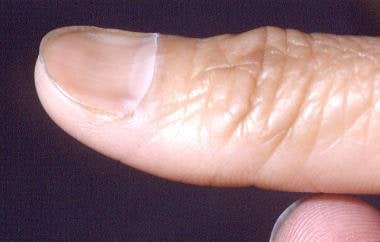
In iron deficiency anemia, reduced oxygen availability is believed to diminish disulfide bond formation, which consequently reduces nail-plate pliability. Changes also occur in the oral mucosa and on the tongue. Of 378 patients with iron deficiency, 14% had angular stomatitis and almost half had alterations in tongue papillae, either a change to the filiform type or atrophy. [25, 26]
Diffuse hair thinning is reported in female blood donors and is believed to be caused by a decrease in the iron storage pool. Changes in hair quality, primarily increased splitting, dryness, and dullness, are infrequently observed in individuals with iron deficiency. Changes in hair quality are related to impairment in keratin production.
Notably, iron deficiency anemia has been observed in patients undergoing extracorporeal photopheresis for cutaneous T-cell lymphoma. [27, 28]
Pernicious anemia
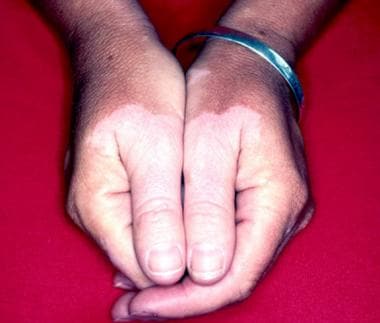
No distinct cutaneous findings occur in pernicious anemia. [29] The deep-red, cobblestone-appearing tongue may be a late manifestation. With excess production of unconjugated bilirubin, the skin tends to have a yellowish hue. Whereas autoimmune phenomena tend to occur together, pernicious anemia may be associated with autoimmune thyroiditis, hypoparathyroidism, and Addison disease. Late-onset vitiligo is reported in as many as 10% of patients with pernicious anemia (see the image below).
Changes in pigmentation of skin appendages have been described. [30] In particular, reversible facial and acral hyperpigmentation has been noted in multiple patients. [31, 32] Some Latin American patients displayed reddish hair discoloration that resolved upon the cobalamin level returning to normal. In 2 African American patients with pernicious anemia, blue nail discoloration was noted.
Sickle cell anemia
Sickle cell anemia, severe alpha and beta thalassemias, hereditary spherocytosis, and pyruvate kinase deficiency may all be complicated by the occurrence of leg ulcerations. Ulcers, usually in the ankle area over the medial or lateral malleoli, are the most common cutaneous symptom in sickle cell anemia. Cutaneous ulcerations occur most commonly in sickle cell disease. They are less common in sickle cell-beta (0) thalassemia and sickle cell-alpha thalassemia, and they are nonexistent in sickle cell hemoglobin C disease and sickle cell-beta (+) thalassemia.
Estimates of the incidence vary, ranging from 75% in Jamaica to 0% in Saudi Arabia. [33] According to the Cooperative Study of Sickle Cell Disease, approximately 25% of patients with sickle cell disease in the United States have a history of active ulcers or developed ulcers during the 8 years of observation. The incidence is low in children younger than 10 years because of the protective effect of persistent fetal hemoglobin. Other risk factors for developing leg ulceration are anemia, male sex, combined HLA-B35 and HLA-Cw4, and antithrombin III deficiency.
Based on clinical experience and epidemiologic studies, the causes of leg ulceration can be divided into 3 categories: reduction in blood supply to the skin (infarction being the primary cause), local edema, and minor trauma. The size of the ulcers varies from a few millimeters to large circumferential lesions. The onset of ulcers is often spontaneous, with prodromal pain or dysesthesias in the area.
A history of previous leg ulcers is the strongest predictor of recurrence. A history of an insect bite or the use of a foot or an ankle for an intravenous site may exist. Secondary infections, mostly with Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus species, and Bacteroides species are most common. Systemic infections, osteomyelitis, and tetanus are rare complications. [34] The leg ulcers are resistant to healing and tend to be recurrent in well over half the patients; additionally, they may be associated with venous incompetence. [34] Pain (often severe), loss of mobility with associated loss of work time, and depression are the primary sources of morbidity. Wearing properly fitting shoes, using insect repellents, and avoiding drawing blood from the feet can prevent ulceration. Encourage patients with a history of ulcers and edema to wear elastic support stockings.
Therapy for leg ulcers involves several modalities, including gentle debridement, control of local edema (strict bed rest and elevation of the leg), treatment of infection, and correction of deficiencies in nutrients (zinc sulfate 220 mg given orally 3 times a day). Although never proven by a controlled study, transfusions to hemoglobin levels greater than 10 g/dL are effective in therapy for resistant ulcers, probably as a way to reduce the hemoglobin S level. Elevation of the fetal hemoglobin level can be achieved by using hydroxyurea, alone or in combination with erythropoietin. Improvement of leg ulcers was also reported in 2 patients given arginine butyrate by means of a similar mechanism. Pentoxifylline has been used on many occasions in patients with leg ulcerations, but the evidence for efficacy in sickle cell disease is lacking. Skin grafts are advocated for ulcers resistant to more conservative therapy.
Hydroxyurea treatment of sickle cell anemia in children can be associated with nail hyperpigmentation, longitudinal bands, and hyperpigmentation of the palms. [35] Such changes have been described after 6-16 weeks of hydroxyurea therapy.
Fanconi anemia
Fanconi anemia is an autosomal recessive disease that involves congenital abnormalities, bone marrow failure, and predisposition to malignancy. [36] Patients have an increased incidence of spontaneous chromosomal abnormalities, due to problems with DNA repair. [37] By the end of childhood, patients with Fanconi anemia develop hypoplastic bone marrow that affects all 3 lineages.
Skin findings consist of several abnormalities of pigmentation. Many patients present with café au lait spots, which, in most individuals, are present at birth. Diffuse hyperpigmentation, which can also be an acquired phenomenon because of iron overload accompanying multiple transfusions, is present early in life. Patients with Fanconi anemia can exhibit hypopigmented macules.
In an interesting pediatric case, 2 Indian brothers with Fanconi anemia presented without café au lait spots, mucosal pigment changes, or palmoplantar keratoderma. The patients had diffuse, discrete hypopigmented mottling over their entire bodies. [38]
A number of malignancies have been reported in patients with Fanconi anemia, particularly myelogenous leukemia. Many individuals die of aplastic anemia. Bone marrow transplantation has been used successfully in this setting.
Plasma-Cell Disorders and Dysproteinemias
Diseases associated with monoclonal immunoglobulin or light-chain production can cause skin findings. Skin lesions can be directly related to the abnormal protein production (acting as cold agglutinins or cryoglobulins or causing an increase in viscosity). Skin diseases (eg, cutaneous plasmacytoma or polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes [POEMS] syndrome) can be observed with clonal plasma cell proliferation. Light-chain deposition in the skin can be a part of the spectrum of symptoms of amyloidosis.
Cryoglobulinemia
Cryoglobulins are serum immunoglobulins complexed with other immunoglobulins or proteins that reversibly precipitate in cold temperatures. Cryoglobulinemia is associated with a range of clinical findings, including palpable purpura in parts of the body exposed to cold temperatures, Raynaud phenomenon, acral hemorrhagic necrosis, arthralgia, neurologic manifestations, and glomerulonephritis.
The classification of cryoglobulinemia follows the initial description by Brouet et al and is based on the involved immunoglobulin. [39] Type I cryoglobulins are monoclonal immunoglobulins, usually IgG or immunoglobulin M (IgM). Type I cryoglobulins account for approximately 5-25% of cases. Patients usually have an underlying B-cell malignancy, such as chronic lymphatic leukemia (CLL), multiple myeloma, or B-cell-type non-Hodgkin lymphoma (NHL). Type I cryoglobulinemia predominantly affects the skin, kidneys, and bone marrow. Cutaneous manifestations can precede extracutaneous manifestations by decades.
Mixed cryoglobulinemias consist of type II and type III cryoglobulins. [40] Type II cryoglobulins are mixed cryoglobulins; 1 component is monoclonal, and 1 component is polyclonal. Type II cryoglobulinemia accounts for 40-60% of cases. Type II cryoglobulins can be associated either with B-cell malignancies or with autoimmune disorders. They are frequently due to persistent viral infections such as hepatitis C and HIV infection. Type III cryoglobulins are polyclonal immunoglobulins that form a cryoprecipitate with polyclonal IgG or a nonimmunoglobulin serum component. Type III cryoglobulinemia can be classified as an immune complex disease. Type III cryoglobulinemia accounts for 40-50% of cases and is not associated with any particular hematologic disorder. Patients with this type of cryoglobulin have an autoimmune process or an infection (eg, with hepatitis B, hepatitis C, or Epstein-Barr virus). [41]
The severity of the symptoms reflects the serum concentration and the temperature at which cryoglobulins precipitate. The most typical clinical presentation of a patient with cryoglobulinemia is the appearance of purpura. Cold sensitivity is present in less than 50% of patients with cryoglobulinemia (more variable than this in type II and III cryoglobulins). [42] Organs and systems usually affected in cryoglobulinemia include the skin, kidneys, and nervous system.
Patients with type I cryoglobulins typically present with cutaneous manifestation related to hyperviscosity and/or thrombosis. In 90-95%, lesions associated with type I cryoglobulinemia consist of erythematous macules to purpuric papules of the lower extremities. Infarction, hemorrhagic crusts, and ulcerations can occur in 10-25% of patients. Raynaud phenomenon, livedo reticularis, and acrocyanosis are common in type I cryoglobulinemia. Cutaneous lesions commonly manifest on the head, neck, and mucosal surfaces. Severe untreated cases can progress to gangrene requiring surgical amputation.
Type II and type III cryoglobulins are associated with arthralgias and vascular purpura, and they can also cause renal and neurologic symptoms. Leukocytoclastic vasculitis, appearing as palpable purpura, is a characteristic lesion of cryoglobulinemia types II and III. The lesions are nonpruritic and transient. Mixed cryoglobulinemias present with the classic Melzer triad of purpura, arthralgias, and weakness in 25-30% of patients. Other cutaneous manifestations include polyarteritis nodosa–like lesions, splinter hemorrhages, and palmar erythema. [42]
Laboratory detection and analysis of monoclonal immunoglobulins or light chains are performed by using serum or urine protein electrophoresis and immunofixation. After serum is separated, it is stored at 4°C. After approximately 24 hours, the cryoprecipitate may undergo immunoelectrophoresis, which defines its composition. The concentration of cryoglobulin may be expressed as cryocrit or in milligrams per milliliter. Type I cryoglobulins are usually present in the highest concentrations (up to 30 mg/mL).
Skin lesions in mixed cryoglobulinemia most often reveal leukocytoclastic vasculitis (50%) and, less commonly, inflammatory or noninflammatory purpura (10-20%), noninflammatory hyaline thrombosis (10%), or postinflammatory sequelae (10%). Direct immunofluorescence microscopy of acute lesions often reveals deposits of IgM, IgG, and/or C3 complement. In contrast, type I cryoglobulinemia more often induces noninflammatory thrombotic lesions, sometimes with evidence of cutaneous infarction or hemorrhage. [43]
Patients with types I or II cryoglobulins should be evaluated for an underlying lymphoproliferative disorder. Type II or III cryoglobulin detection should prompt the search for connective-tissue and/or autoimmune-type disorders or chronic infection.
Treatment of cryoglobulinemia is based on the severity of the clinical presentation. Mild cutaneous symptoms may call for only avoidance of exposure to cold temperatures. Nonsteroidal anti-inflammatory drugs (NSAIDs) can alleviate bothersome dermatologic and articular manifestations. Renal involvement may require corticosteroids and/or cytotoxic agents. Plasmapheresis has been used in progressive refractory disease, but it must be combined with cytotoxic agents to avoid postpheresis rebound.
Patients with hepatitis C–associated mixed cryoglobulinemia should undergo treatment with pegylated interferon-alfa and ribavirin if clinically appropriate. Anti-CD20 (rituximab [Rituxan]) and anti-TNF (infliximab [Remicade]) have also been used successfully to treat hepatitis C–associated mixed cryoglobulinemia when the therapy described above is ineffective, contraindicated, or intolerable.
Hyperviscosity syndrome
Hyperviscosity syndrome is caused by a clinically significant increase in the viscosity of whole blood, which may result from an increased number of cellular blood elements, as in myeloproliferative disorders (eg, polycythemia vera, essential thrombocythemia), or a large amount (>3 g/dL) of monoclonal proteins (mostly the IgM type) in the blood. In clinical terms, hyperviscosity syndrome may manifest as mucous membrane bleeding, retinopathy, cardiac failure, and neurologic disturbances.
If hyperviscosity syndrome is present because of cryoglobulins, Raynaud phenomenon can be observed. For symptoms to develop, blood viscosity must increase at least 4-fold. A bleeding tendency can also be explained by clotting-factor antibodies and/or platelet dysfunction because of surface coating by immunoglobulins. Plasma exchange should be started in patients with severe neurologic impairment. Discontinuation of plasma exchange results in recurrence of symptoms in 1-3 weeks if chemotherapy directed to the underlying dysglobulinemia is not administered.
Cold agglutinin disease
Cold agglutinins are antibodies, mostly the IgM type, that are directed against the I/i antigens on the surface of RBCs. Cold agglutinins are able to agglutinate RBCs, usually below the body temperature. Anti-I antibodies primarily occur with idiopathic disease, with some lymphomas, and with mycoplasmal pneumonia. Anti-I–specific antibodies are associated with infectious mononucleosis and with some lymphomas.
Cold agglutinins can be divided into 2 types: monoclonal, which occurs in the idiopathic form or which is associated with B-cell proliferation, and polyclonal, which accompanies infectious diseases, especially Mycoplasma pneumoniae infection. Cold agglutinin disease manifests as episodes of hemolytic anemia, hemoglobinuria, and cold-provoked vaso-occlusive phenomena. The most common cutaneous manifestations associated with cold agglutinins are acrocyanosis and Raynaud phenomenon. Livedo reticularis, cold-induced urticaria, petechiae, ecchymoses, and hemorrhagic bullae have also been described. Skin ulceration or necrosis is unusual. Jaundice and/or pallor can follow a severe episode of hemolysis.
Acral areas (eg, earlobes, nose, distal extremities) are preferentially affected, most likely because of lower temperatures at these sites. Symptoms associated with agglutination are quickly reversible on warming. The severity of the clinical manifestation does not appear to be related to the agglutinin titer. On microscopic evaluation, dilatation of dermal vessels is extensive, with thinning of vascular walls and without evidence of vasculitis. Eosinophilic thrombi or fibrin can occlude some capillaries.
In patients with small-vessel thrombosis induced by cold agglutinin disease, the differential diagnoses must include disseminated intravascular coagulation (DIC), thrombotic thrombocytopenic purpura (TTP), warfarin (Coumadin)–induced skin necrosis, cryofibrinogenemia, and paroxysmal nocturnal hemolysis (PNH).
The best treatment of cold agglutinin disease is avoidance of cold temperatures. In contrast to the treatment of other hemolytic anemias, steroids and splenectomy have no role in the treatment of hemolytic anemia due to cold agglutinins. Cytotoxic agents, such as cyclophosphamide and chlorambucil, may be useful in selected patients to reduce the production of antibody. Anti-CD20 (rituximab [Rituxan]) has been effective in treating patients with cold agglutinin disease that did not respond to conventional therapies. Plasmapheresis should be reserved for patients with ongoing severe hemolysis, severe acrocyanosis, or anti-Pr antibodies and polyneuropathy, and as preoperative management in select cases. Plasmapheresis was successfully used in certain clinical scenarios, but it is generally avoided because of the risk of inducing hemolysis. When clinically indicated, plasmapheresis should be performed with equipment, lines, and solutions that have been warmed to prevent hemolysis.
Plasmacytoma
Neoplasms originating from mature secretory B lymphocytes (plasma cells) can occur as a spectrum of diseases, ranging from solitary or multiple plasmacytomas to multiple myeloma. Multiple myeloma arises from bone-marrow plasma cells and typically manifests as osteolytic lesions on the skull, vertebrae, ribs, pelvis, and proximal long bones. Anemia, kidney dysfunction, hypercalcemia, and susceptibility to bacterial infections frequently accompany multiple myeloma. Monoclonal peaks of immunoglobulin are found on electrophoresis and occasionally accompanied by light chains in the urine.
Several nonspecific lesions, including amyloidosis, cold urticaria, alopecia, pigmentation, and Raynaud phenomenon, are associated with multiple myeloma. Follicular hyperkeratosis, most prominent on the nose, is considered a sign of paraneoplastic myeloma.
Cutaneous plasmacytoma of the skin manifests as nontender, smooth, cutaneous or subcutaneous nodules 1-5 cm in diameter. The lesions are flesh- or plum-colored and can become crusted or ulcerated. The lesions are mostly distributed on the extremities, trunk, and face. Most often, the lesions manifest as secondary cutaneous plasmacytomas in the setting of multiple myeloma. Lesions may develop as a direct extension of underlying bone lesions, or, more frequently, they manifest as metastatic foci. Primary cutaneous plasmacytomas with no evidence of involvement of other tissues are exceedingly rare.
Upon histologic analysis, both the dermis and the subcutaneous tissues are infiltrated by plasma cells that vary in maturity. Plasma cells can be distinguished from benign plasma cell proliferation (cutaneous plasmacytosis) on the basis of their monoclonality (presence of only kappa light chains or only lambda light chains). Immunoglobulins produced may be of any type, but immunoglobulin A (IgA) appears to be frequently found.
Therapy for cutaneous plasmacytomas in the setting of multiple myelomas includes chemotherapy and local radiation therapy. Surgical excision has a role in lesions limited to the skin that are resistant to radiation therapy.
POEMS syndrome
POEMS syndrome was described as a separate pathologic entity in Japan (on the basis of a series of 102 patients). Since 1968, a few cases have been reported in the United States. Of the originally reported patients, 75% had a serum and occasionally a urine monoclonal spike, and, in 95%, the monoclonal protein had a lambda light chain restriction. In the Japanese series, men were affected twice as often as women, with a mean age of 46 years. Polyneuropathy was present in all patients and was usually sensorimotor in nature. Organomegaly manifested mostly as hepatomegaly (82%), lymphadenopathy (65%), or splenomegaly (39%). Endocrinopathy occurred as impotence (78%) or gynecomastia (68%) in men and as amenorrhea (68%) in women. Other endocrine abnormalities were glucose intolerance, hyperthyroidism, hyperprolactinemia, and adrenal insufficiency.
In contrast to multiple myeloma, no anemia, no renal insufficiency, and only rare cases of hypercalcemia are reported. Bone marrow infiltration by plasma cells is unusual. Patients have bony lesions, but unlike those in multiple myeloma, the bony lesions are mostly osteosclerotic.
Skin changes are commonly observed in patients with POEMS syndrome, with 93-98% having diffuse hyperpigmentation and 92% having peripheral edema. Skin thickening with sclerodermoid changes and thickening, which can limit function, are seen in 77% of patients. Ascites and pleural effusions are occasionally reported. Hypertrichosis, manifested as coarse black hair, appears on extremities in one fourth of patients. Hypertrichosis is either generalized or limited to certain body areas, such as extremities or face. Digital clubbing, white fingernails, sicca syndrome, acrocyanosis (20%), plethora (20%), hemangiomas (10%), telangiectasias (10%), and Raynaud phenomenon are also described. [44]
Solitary osteosclerotic lesions can be treated with local radiation in a dose of 40-50 Gy. Systemic and skin changes tend to respond within 1 month and can continue to respond as long as 2-3 years later. Widespread systemic disease requires treatment with systemic chemotherapy. Melphalan and prednisone were given in combination to 48 patients with POEMS of which 44% had improvement. [45] Lenalidomide has also produced encouraging results. [46] Autologous hematopoietic stem cell transplantation has been used successfully in young patients after high-dose melphalan conditioning chemotherapy.
Amyloidosis
Amyloidosis consists of a group of disorders in which amyloid fibrils are extracellularly deposited in internal organs, including the skin. With time, the amyloid fibrils destroy the healthy ultrastructure of the organs, affecting their function. On x-ray diffraction studies, amyloid fibers demonstrate a beta-pleated structure, unlike that of most human proteins (which are synthesized in an alpha-helical structure). The proteins that can serve as precursors for fibrils can be either light-chain monoclonal proteins or serum protein A. This distinction serves as a basis for classification into amyloid light-chain amyloidosis (AL) and serum protein A amyloidosis (AA).
AA-type amyloidosis (secondary systemic amyloidosis) is usually associated with a chronic inflammatory process, such as rheumatoid arthritis, chronic osteomyelitis, tuberculosis, or leprosy. Although the deposition of amyloid in the skin is common in AA, this deposition rarely leads to clinically apparent skin lesions.
AL-type amyloidosis is the primary form and includes forms associated with multiple myeloma and Waldenström macroglobulinemia. AL commonly affects the skin, with a reported incidence of 21-40%. The most characteristic skin lesions are nonpruritic, nontender, shiny, waxy papules, which commonly develop on the eyelids, also know as raccoon eyes. Lesions can also be present in the skin folds, the retroauricular folds, the anogenital region, or the oral mucosa. Subcutaneous nodules or plaques are also described.
Purpura is the type of lesion encountered most often because of the frequent deposition of amyloid in blood vessel walls, which results in the extreme fragility of skin vessels. The deposition of amyloid in the blood vessel walls is also responsible for pinch purpura, which is frequently found on the eyelids of patients with AL-type amyloidosis. Scalp involvement may be evident with hair loss. Mucocutaneous changes in the oral cavity can include rubbery papules, petechiae, and ecchymoses
AL amyloid deposition is also responsible for peripheral neuropathy, carpal tunnel syndrome, orthostatic hypotension (autonomic neuropathy), congestive heart failure, and nephrotic syndrome. Approximately 10% of patients with AL amyloidosis (usually with advanced disease) develop macroglossia, which can produce dysphonia and dysphagia. Another manifestation of oral amyloidosis may be xerostomia, resulting from salivary gland infiltration by amyloid.
Primary localized cutaneous amyloidosis does not appear to be related to systemic illness. The papular form, often referred to as lichen amyloidosis, manifests with extremely pruritic, hyperkeratotic, brown papules. The papules are most commonly the size of a pinhead, but they can reach 6-8 mm in diameter and usually occur on the shins. Macular amyloidosis is probably a variant of lichen amyloidosis, manifesting as pruritic, oval, grayish-brown macules on the lower extremities or the back. Biopsy samples of tissue with lichen amyloidosis and macular amyloidosis differ from samples of tissue with systemic amyloidosis because of sparing of dermal blood vessels.
Definite diagnosis of systemic amyloidosis requires the confirmation of amyloid deposits in the tissue. The most common sites include the rectum and the tongue; however, several reports in the literature state that skin biopsy is a sensitive and comparatively safe and uncomplicated procedure. Evaluation of the patient with biopsy-proven cutaneous amyloid deposition depends on the clinical scenario. Vessel-wall infiltration and epidermal atrophy in a biopsy sample obtained from a patient with waxy eyelid nodules should suggest AL-type amyloidosis, and a search for underlying multiple myeloma or Waldenström macroglobulinemia is warranted. If all clinical and pathologic features appear to suggest lichen amyloidosis, no further search is necessary, and local measures should be undertaken.
Effective means of treating AL- and AA-type amyloidosis are not yet available, and, most of the time, treatment is symptomatic. AA-amyloid treatment should be focused on treating the underlying disease. Patients with hereditary amyloidosis have successfully undergone liver transplantation with regression of symptoms and a decrease in amyloid burden. As an alternative, patients with severe end-organ damage have received combined hepatorenal and hepatocardiac transplantation.
Mastocytosis
Mastocytosis is a disorder of mast cell proliferation that has both cutaneous features and systemic features. The most frequent site is the skin, although mast cell hyperplasia can occur in the bone marrow, liver, spleen, lymph nodes, GI tract, and skeletal system (see the images below).
Mastocytosis is divided into distinct clinicopathologic entities based on clinical presentation, pathologic findings, and prognosis.
The following World Health Organization (WHO) classification represents the current clinical approach to distinguishing among the different disorders:
-
Indolent mastocytosis: This includes smoldering systemic mastocytosis and isolated bone marrow mastocytosis.
-
Systemic mastocytosis: This occurs with an associated hematologic non–mast cell lineage disorder.
-
Aggressive systemic mastocytosis: Patients have lymphadenopathy with blood or tissue eosinophilia.
-
Mast cell leukemia: Patients are grouped as those with more than or those with less than 10% mast cells in the peripheral blood.
-
Mast cell sarcoma
-
Extracutaneous mastocytoma
Patients whose conditions fall into the indolent category have a good prognosis, whereas patients with all other disorders have a relatively poor prognosis.
Human mast cells originate from pluripotent bone marrow cells and then circulate to specific body sites, where they undergo maturation into fully granulated cells. The skin and the GI tract are particularly rich in mast cells. In both lesional skin and healthy skin of patients with urticaria pigmentosa, the expression of the soluble form of stem cell factor is increased. Lesional skin of most adult patients with sporadic cutaneous mastocytosis contains a mutated form of c-kit, the gene encoding the mast cell growth factor receptor.
The clinical symptoms of mastocytosis are due to increased production of mast cell mediators. The clinical symptoms can be grouped into 3 categories: granule associated (eg, histamine, heparin, tryptase), lipid derived (eg, leukotrienes, prostaglandin D2, platelet-activating factor), and cytokines (eg, transforming growth factor [TGF]-beta TNF-alpha, interleukin [IL])–3, IL-5). Rubbing the lesion causes the development of a wheal and erythema (Darier sign). Various substances (eg, alcohol, dextran, morphine, codeine) can exacerbate symptoms by degranulating the mast cells. Diarrhea, syncope, headache, and wheezing occasionally accompany flushing.
Skin lesions include localized lesions (eg, mastocytoma) and generalized cutaneous mastocytosis (eg, urticaria pigmentosa, telangiectasia macularis eruptiva perstans, diffuse cutaneous mastocytosis).
Although mastocytomas may be present at birth, they mostly appear within the first 3 months of life; they are rarely described in adults. Lesions are solitary or multiple but few in number. They appear as macules or papules and are yellowish to tan.
In urticaria pigmentosa, lesions occur in most patients with indolent mastocytosis and in less than half the patients with mastocytosis with an associated hematologic disorder. Lesions of urticaria pigmentosa are characterized by small yellow-tan to reddish-brown macules or slightly raised papules. [47] Nodules or plaquelike lesions can occur. The lesions are randomly distributed, often sparing the face, feet, and soles (see the images below).
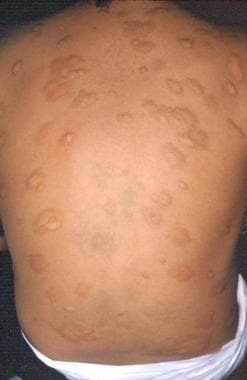
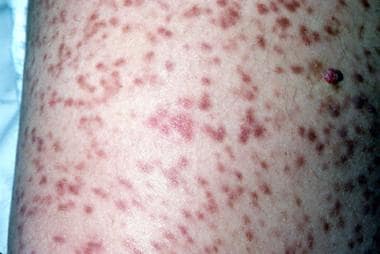
The Darier sign of urticaria pigmentosa is characterized by urticaria and erythema upon rubbing, scratching, or stroking the affected skin. Pruritus associated with urticaria pigmentosa may be exacerbated by changes in the temperature, exercise, hot showers, spicy foods, ethanol, or certain drugs. [52] Bullous eruptions with hemorrhages can also occur in children. In various studies, 10-70% of adult patients with urticaria pigmentosa had systemic disease. Of infancy-onset lesions, 50% of the lesions resolve by the time the patient is an adolescent. The remainder of the lesions lighten in color (see the image below).
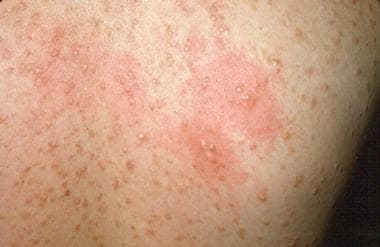
Telangiectasia macularis eruptiva perstans is a rare form of mastocytosis that occurs mostly in adults. Lesions are 2-6 mm in diameter tan-to-brown macules with telangiectasias. [53] They become swollen when rubbed, but they are not pruritic or bullous. The amount of mast cells around the capillaries and venules of the superficial vascular plexus are increased.
In patients with diffuse cutaneous mastocytosis, the skin may have a healthy appearance or a peau d'orange appearance. Large bullae may occur spontaneously or following trauma. The bullae may subsequently rupture, forming erosions. Eruptions can be spontaneous or can be triggered by infection or vaccination. Because blisters can be present at birth, cutaneous manifestations are a diagnostic possibility among individuals with neonatal blistering disease. Children are at risk for flushing, hypotension, shock, and death.
Differential diagnoses include carcinoid syndrome for diffuse cutaneous mastocytosis-induced flushing, juvenile xanthogranuloma, and Spitz nevus for mastocytoma. Urticaria pigmentosa and telangiectasia macularis eruptiva perstans should be differentiated from histiocytosis X, secondary syphilis, papular sarcoid, and general eruptive histiocytoma. Patients with any form of mastocytosis should avoid triggers, such as friction, exertion, alcohol ingestion, medications (eg, NSAIDs, opioids), or temperature changes.
No therapy is available to change the occurrence or natural course of urticaria pigmentosa or cutaneous mastocytosis. Therapy is intended to prevent mast cell mediator release and to treat symptoms when they arise. Solitary lesions that do not undergo involution may be surgically removed. Diffuse cutaneous mastocytosis usually resolves spontaneously when patients are younger than 5 years. Childhood urticaria pigmentosa resolves in half the patients. Patients with urticaria pigmentosa are initially treated with oral anti-H1 antihistamines. The combined use of H1 and H2 antihistamine antagonists and disodium cromoglycate reduces the pruritus and whealing of urticaria pigmentosa.
In extensive cutaneous disease unresponsive to other forms of treatment, oral psoralen UV-A (PUVA) photochemotherapy can be used, which can decrease the pruritus and whealing of urticaria pigmentosa in adults, but relapses within 3-6 months after cessation of therapy are common. Oral PUVA photochemotherapy diminishes pruritus and bullae formation in pediatric patients with diffuse cutaneous mastocytosis. Systemic corticosteroids have been used without notable effect, but the application of potent topical glucocorticoid 0.05% betamethasone dipropionate ointment under plastic film occlusion for 8 hours daily for 6 weeks resolves both the pruritus and the Darier sign in patients with urticaria pigmentosa.
Aggressive systemic mastocytosis is commonly treated with interferon alfa-2b and/or glucocorticoids or cladribine. Mast cell leukemia is treated similarly to patients with acute leukemia. Even with aggressive chemotherapy, patients with mast cell leukemia have a poor prognosis, and bone marrow transplantation can be considered.
Improved understanding of the underlying molecular mechanism may soon lead to novel therapeutic approaches. Some forms of mastocytosis are caused by c-kit mutations, which constitutively activate kit kinase. Imatinib (Gleevec) has been effective in patients with mutant c-kit. Disease associated with c-kit D816V mutation is resistant to imatinib and warrants further investigation with newer agents.
Myeloproliferative Disorders
Polycythemia vera
An absolute increase of circulating RBCs that causes an increase in hematocrit values higher than 55% characterizes polycythemia vera. Polycythemia vera can be accompanied by a moderate increase in leukocytes and, less frequently, by thrombocytosis. Skin changes are characteristic. Ruddiness of the complexion and occasionally cyanosis, affecting mostly the face, neck, and distal parts of the extremities, may occur. Less frequent changes are telangiectasias and bleeding gums. Rosacea and purpura can be present.
Pruritus, which is triggered by a bath or a shower and which lasts up to 1 hour, is a typical symptom in patients with polycythemia vera. The degree of pruritus is not related to the severity of the disease. Use of antihistamines (H1 and H2 blockers) is indicated because elevation of the histamine levels corresponds with symptoms. However, failure of the pruritus to respond to antihistamine therapy in many patients suggests that an increased histamine level is not the sole factor in symptom development. Patients with polycythemia vera often have iron deficiency, which has been implicated as a contributing factor. Anecdotal reports describe the use of selective serotonin reuptake inhibitors (SSRIs) in patients with polycythemia vera in whom pruritus responded favorably. [54]
Polycythemia vera and essential thrombocythemia, among other disorders, can be the underlying causes of erythromelalgia (a form of paroxysmal vasodilatation affecting the skin), which manifests with an intense burning sensation, pain, and increased local skin temperature. These paroxysms may last minutes to days and may be relieved by cooling. They are rapidly reversed with antiplatelet therapy. This fact, and the preservation of arterial pulses, appear to indicate that abnormal arachidonic acid metabolism occurs in the platelets in persons with polycythemia vera. Erythromelalgia responds well to low-dose aspirin 50-100 mg/d and to a reduction in the platelet count. Livedo reticularis is also described. [55]
The mainstay of treatment for polycythemia vera is to maintain the hematocrit below 45% (0.45) in men and 42% (0.42) in women. Phlebotomy alone is effective, but the risk of thrombosis approaches 25% in the first 2 years of treatment. Patients at high risk for thrombosis (age >70 y, previous thrombosis, other cardiovascular risk factors, platelet count >1500 X 109/L [>1500 X 103/µL) should receive supplemental hydroxyurea or anagrelide. Anagrelide is associated with an acquired idiopathic cardiomyopathy and must be used with extreme caution in patients with known or suspected heart disease. [56, 57] Low-dose aspirin 75-100 mg/d should be given to all patients if not contraindicated. Interferon-alfa can be used in patients with refractory pruritus, in high-risk women of childbearing potential, and in patients whose condition is refractory to all other treatments.
Investigations have identified a single point mutation (V617F) in the tyrosine kinase gene Janus kinase 2 (JAK2) on the short arm of chromosome 9 in certain myeloproliferative disorders. In 1 study, JAK2 was identified in 81% of patients with polycythemia vera and in 43% of patients with essential thrombocythemia. [58] Tyrosine kinase dysregulation may suggest a possible role for tyrosine kinase inhibition similar to that seen with imatinib mesylate (Gleevec) in chronic myeloid leukemia (CML).
Cutaneous myelofibrosis
Cutaneous myelofibrosis is a myeloproliferative disorder with a clonal proliferation of defective stem cells in the bone marrow. The hallmark of myelofibrosis is extramedullary hematopoiesis, which rarely presents as a cutaneous form. Overproduction of abnormal megakaryocytes produces excess amounts of platelet-derived growth factor, which is a stimulus for fibroblast proliferation and collagen production. Although involvement of the skin is rare, it may manifest as erythematous plaques, nodules, erythema, ulcers, or bullae. Microscopy of cutaneous lesions (dermal and subcutaneous nodules) shows infiltrates of mature and immature myeloid cells, erythroid precursors, and a predominance of megakaryocytes. Marked production of reticulin fibers is typical. Neutrophilic dermatoses can be a presenting or a complicating feature of myelofibrosis, and it can progress to bullae or pyoderma gangrenosum.
Hydroxyurea-induced skin changes
Hydroxyurea is probably the medication most commonly used to treat polycythemia vera and essential thrombocythemia. Salmon-Ehr et al prospectively followed 26 patients treated with hydroxyurea over 2 years. [59] All patients except 1 developed skin-related disorders during the follow-up period. The most frequent skin disorders were skin dryness (16 patients), melanonychia (8 patients), actinic keratosis (8 patients), leg ulcers (8 patients), increased skin pigmentation (5 patients), and squamous cell cancer (2 patients). Other manifestations include alopecia (rare), facial and peripheral edema, gangrene, nail and/or skin atrophy, scaling, and violet papules.
Leukemia
Overview
In leukemia, specific or nonspecific lesions of the skin may occur. Specific lesions (leukemia cutis) contain leukemia cells, which directly infiltrate the epidermis, dermis, or subcutaneous fat. Nonspecific lesions, which are more common, are considered a cutaneous reaction to the ongoing malignancy process.
Specific lesions
Leukemia cutis includes granulocytic sarcoma, chloroma, and myeloblastoma and represents localized or disseminated skin infiltration by blast cells. Usually, leukemia cutis is a sign of relapse or dissemination of medullary disease. Overall, leukemia cutis is uncommon; however, it is most common in people older than 50 years. Most often, leukemia cutis is associated with acute monocytic leukemia and acute myelomonocytic leukemia, but it can be observed in any acute or chronic leukemia, including the leukemic phase of non-Hodgkin lymphoma and hairy cell leukemia.
Skin pain, tenderness, and pruritus are usually absent. Most commonly, lesions are discrete, firm, violaceous or red-brown papules, nodules, or plaques. On occasion, ulcerative lesions or hemorrhagic bullae are observed.
The differential diagnoses include bacterial, viral, and fungal dermatoses; drug eruptions; leukocytoclastic vasculitides; Sweet syndrome; and pyoderma gangrenosum. Leukemia cutis may occur simultaneously with or before the medullary disease. Leukemia cutis typically develops late in the course of established acute leukemia. In contrast, when it develops in association with chronic myelogenous leukemia or other myeloproliferative disorders, leukemia cutis is usually a sign of impending blastic transformation. Early cutaneous involvement occurs more frequently in acute myelomonocytic leukemia and monocytic leukemia. Leukemia cutis is strongly associated with extramedullary involvement of other sites, including the meninges.
The histopathologic diagnosis of leukemia cutis rests on the demonstration of a diffuse and infiltrative population of mononuclear cells, which are accompanied by granulocytic cells at various stages of maturation and typically include eosinophilic myelocytes. To establish the diagnosis of leukemia cutis in skin biopsy specimens, chloroacetate esterase (CAE) activity; Leu-M1 positivity; and the presence of lysozyme, alfa1-antitrypsin, and alfa1-antichymotrypsin serve as supportive findings, with lysozyme being the most consistent positive marker.
The prognosis for leukemia cutis is related directly to the prognosis for the systemic disease. Therapy is usually directed at the underlying leukemia. Systemic chemotherapy sufficient for the bone marrow remission may not effectively treat the cutaneous lesions. A combination of systemic chemotherapy and local electron-beam therapy may be necessary. The radiosensitizing properties of anthracyclines should be kept in mind when used before or in conjunction with electron-beam irradiation.
Nonspecific (reactive or secondary) lesions
Nonspecific (reactive or secondary) lesions are occasionally referred to as paraneoplastic leukemia–associated syndromes. Nonspecific lesions are common and encompass infections of the skin, especially in the setting of neutropenia, cutaneous vasculitis, or neutrophilic dermatoses, and are histologically characterized by granulocyte infiltration without malignant cells in the biopsy specimen. Sweet syndrome, pyoderma gangrenosum, subcorneal pustular dermatosis of Sneddon-Wilkinson, and erythema elevatum diutinum are examples of neutrophilic dermatoses.
Sweet syndrome
In 1964, Sweet described Sweet syndrome, also known as acute febrile neutrophilic dermatosis (see the images below).
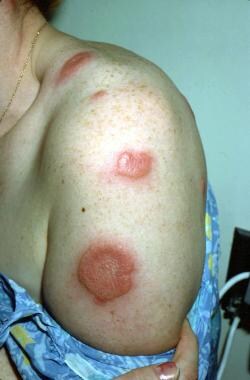
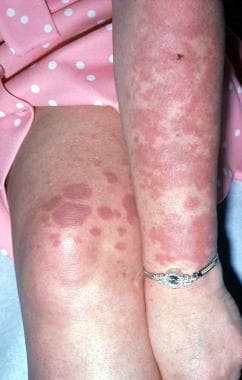
Fever; neutrophil leukocytosis; painful, erythematous, cutaneous plaques; and a dense dermal infiltrate of neutrophils without vasculitis characterize Sweet syndrome. Sweet syndrome is mostly idiopathic, except for 10-20% of cases, which are associated with hematopoietic malignancies (eg, acute myelogenous leukemia [AML], myelodysplastic syndrome, lymphoproliferative disorders, multiple myeloma). Rare cases have been associated with solid malignancies (mostly of genitourinary origin).
Sweet syndrome associated with acute myelogenous leukemia is often the presenting sign of acute myelogenous leukemia. The pathogenesis of Sweet syndrome is unclear; it may represent a type of hypersensitivity reaction to tumor antigens. A high level of granulocyte colony-stimulating factor (G-CSF) and IL-6 in myelodysplastic syndrome is suggested as being responsible for skin manifestations.
The cutaneous eruption consists of erythematous-to-violaceous tender papules, which enlarge to form plaques with an irregular pseudovesicular surface. [60] The skin lesions are usually edematous, beefy, red papules or plaques. In hematologic malignancy–associated Sweet syndrome, bullous lesions may occur. Lesions are reddish and tender. Central clearing may appear. Plaques may cause pain and a burning sensation but are not pruritic. The lesions are most commonly distributed on the face, neck, and upper or lower extremities. Truncal lesions are uncommon. Mucous membranes can be affected, with the appearance of conjunctivitis. Oral lesions, which develop in 3-30% of patients, may be pseudopustular and may ulcerate later. [60] The skin eruptions are usually accompanied by fever, marked erythrocyte sedimentation rate elevation, and leukocytosis.
The differential diagnoses of Sweet syndrome in patients with acute myelogenous leukemia include leukemia cutis, cutaneous and systemic infections, pyoderma gangrenosum, vasculitic syndromes, erythema nodosum, and erythema multiforme. Skin biopsy samples are essential in confirming the diagnosis. Neutrophilic dermal infiltrate and the absence of organism, leukemic infiltration, and vasculitis characterize skin biopsy findings.
The most effective therapy for Sweet syndrome is treatment of the underlying disorder. If the underlying disorder is difficult to discern, steroids are effective. The usual dose is 1 mg/kg, with symptomatic relief occurring within hours. Improvement in lesions is observed within several days. Steroids should be tapered over 4-6 weeks. Abrupt discontinuation or rapid taper of steroids commonly causes a recurrence of lesions. Other drugs reportedly effective in the treatment of Sweet syndrome include colchicine, potassium iodide, and indomethacin.
Pyoderma gangrenosum
Pyoderma gangrenosum is an inflammatory skin disease that occurs in association with systemic disorders in 50-80% of cases in various series (see the images below).
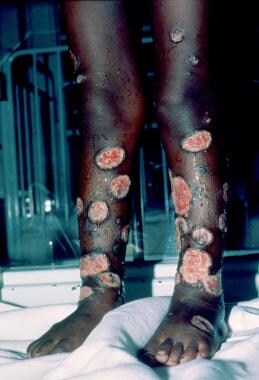
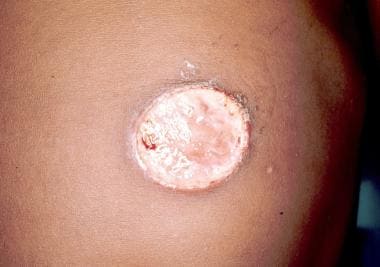
Its most common association is with ulcerative colitis. Approximately 1% of cases of pyoderma gangrenosum occurs in association with hematologic disorders, mostly with myelogenous leukemia but also with polycythemia vera, myelofibrosis with agnogenic myeloid metaplasia, essential thrombocythemia, multiple myeloma, and paraproteinemias. Pyoderma gangrenosum can precede the diagnosis of leukemia or can be part of the initial presentation. If a patient with pyoderma gangrenosum develops acute myelogenous leukemia, the development tends to happen within 1 year of the appearance of the skin changes.
Classic and atypical pyoderma gangrenosum are the 2 main variants of pyoderma gangrenosum. [61] Several other variants may exist. Painful nodules or pustules, which open to form enlarging ulcers with tender raised borders, frequently vesicular or bullous, characterize classic pyoderma gangrenosum. Lesions are most often solitary, but, if multiple, they may coalesce. Systemic symptoms of fatigue and fever may accompany the skin lesions. Lesions are mostly distributed on the lower extremities, but they can also appear on the trunk, abdomen, and, rarely, the head and neck. Atypical pyoderma gangrenosum has a vesiculopustular "juicy" component. [61] This is usually only at the border and is superficially ulcerated. Atypical pyoderma gangrenosum often occurs on the dorsum of the hands, extensor surface of the forearms, or face.
The pathogenesis of pyoderma gangrenosum is unclear. Aberrations of cellular immunity and neutrophilic function have been reported. The histopathologic findings are nonspecific. The findings include ulceration and necrosis of the epidermis and the dermis, chronic inflammatory cells in the center of the lesion, and a dense infiltration of neutrophils at the margins. The diagnosis is strictly clinical. Lesions are typically sterile unless secondary infection occurs.
Pyoderma gangrenosum should be differentiated from bacterial cellulites, herpes simplex virus infection, atypical mycobacterial infection, and deep fungal mycoses. Excluding the infection by using cultures is critical before treatment is instituted.
Corticosteroids remain the treatment of choice. In patients with underlying hematologic malignancies, topical treatment is generally not effective. Systemic doses of up to 60-80 mg/d may be administered. Intravenous pulse methylprednisolone remains an option in refractory cases. Lesion regression with the treatment of the underlying disease has been reported.
Other systemic treatments include dapsone, sulfasalazine (with or without steroids), and clofazimine. Immunosuppressive agents (eg, 6-mercaptopurine, azathioprine, cyclosporine, cyclophosphamide) have been used. High-dose IVIG at 1 g/kg for 2 days is effective in some cases. Split-thickness skin grafting in conjunction with immunosuppressive therapy may be useful for large lesions.
The optimal sequence of therapy (chemotherapy and pyoderma gangrenosum treatment) should be tailored to the patient. On one hand, extensive loss of skin integrity causes patients with pyoderma gangrenosum to be more prone to sepsis at the time of chemotherapy-induced neutropenia. On the other hand, immunosuppression with steroids remains an issue.
Purpura
The term purpura comes from the Greek word porphyra, which is a species of mollusk that was a source of purple dye. Purpura is the outcome of the extravasation of blood into the skin or the mucous membrane, which, in turn, is caused by a defect in primary hemostasis. Primary hemostasis is mediated by platelets, the vessel wall, and the interaction between the platelets and the vessel wall. A multitude of hematologic disorders can result in purpura, and a detailed description of these disorders is not within the scope of this article.
Purpura can be divided as follows into four groups based on the characteristics (eg, size, shape, depth) of blood extravasation:
-
Petechiae are superficial, pinpoint (< 3 mm), red or purple, nonblanching macules that mostly occur in dependent areas. Petechiae imply a platelet-related condition or vessel disease.
-
Ecchymoses are larger than 3 mm, flat, and observed with notable extravasation commonly known as a bruise. Ecchymoses initially form an irregular purple patch, which eventually turns yellow and fades.
-
Vibices are purpuric lesions that are linear and are mostly caused by scratching.
-
Hematomata are deeper collections of blood in the skin. Fluctuation is often palpated.
Underlying reasons for purpura can be divided into several groups, as follows:
-
Thrombocytopenia (most common cause for purpura) and platelet dysfunction
-
Thrombocythemia
-
Vascular disorders (eg, structural vessel malformation, disorders of connective tissue, small-vessel vasculitis)
Thrombocytopenia
Platelets arise from bone marrow and are a result of fragmentation of polyploid megakaryocytes. After leaving the marrow, approximately one third of the platelets are sequestered in the spleen, whereas the rest circulates in the bloodstream for 7-10 days, after which they senesce and are removed by phagocytic cells. The normal blood cell count is maintained at 150-450 X 109/L (150-450 X 103/µL). Petechiae occur at sites of minor trauma when the fully functional platelet count falls below 140 X 109/L (40 X 103/µL), or they can spontaneously appear if the platelet count is lower than 10 X 109/L (10 X 103/µL).
One of 4 mechanisms cause thrombocytopenia: (1) artifactual thrombocytopenia, (2) decreased bone marrow production, (3) increased splenic sequestration, or (4) accelerated destruction of platelets.
Artifactual thrombocytopenia should be considered in patients who have a low platelet count but no petechiae, which occurs in vitro because of an inaccurate count. An inaccurate count can occur because of platelet clumping (pseudothrombocytopenia), the presence of giant platelets, or platelet satellitism (platelet adherence to leukocytes). To exclude platelet count misinterpretation, a review of a blood smear is necessary.
Decreased production of bone marrow is a result of bone marrow injury. Because the injury affects multiple cell lines, thrombocytopenia may be accompanied by leukopenia and anemia. Thiazide diuretics and alcohol have also been implicated. Other common causes of decreased platelet production are replacement of bone marrow by fibrosis or malignant cells, aplastic anemia, vitamin deficiencies, and Wiskott-Aldrich syndrome. Selective impairment of megakaryocyte production, as in congenital megakaryocytic hypoplasia, thrombocytopenia, and absent radii syndrome, rarely occurs. The only way to diagnose the platelet underproduction is with examination of the bone marrow aspirate and the biopsy sample.
Increased platelet sequestration results when more than one third of the produced platelets are sequestered as the spleen increases in size, which, in turn, lowers the platelet count in the bloodstream. The most common causes of splenomegaly include portal hypertension caused by liver disease or splenic infiltration either with tumor cells (eg, in lymphoma) or by macrophages in storage diseases (eg, Gaucher disease). Hypothermia has also been known to cause thrombocytopenia, mostly because of platelet swelling and irreversible aggregation.
Accelerated platelet destruction is the most common cause of thrombocytopenia, which can be caused by either an immunologic process or a nonimmunologic process. Immunologic causes can be further divided into autoimmune and alloimmune; autoimmune being either idiopathic (idiopathic thrombocytopenic purpura [ITP]) or secondary to drugs, infections, pregnancy, or collagen-vascular disorders, and alloimmune being represented by posttransfusion purpura. Nonimmunologic platelet destruction can be caused by thrombotic microangiopathies (eg, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura), infections, or platelet damage via abnormal vascular surfaces (eg, valves, grafts).
Idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura occurs because of the premature destruction of platelets resulting from autoantibody or immune complex deposition on the platelet's membrane. The diagnosis of idiopathic thrombocytopenic purpura remains one of exclusion because currently available assays for platelet-associated antibodies are nonspecific and have poor sensitivity. First, normal (or increased) megakaryocyte count on bone marrow examination and an absence of splenomegaly should be noted. Excluding possible underlying causes (eg, drugs, infection, chronic lymphatic leukemia, lymphoma) is of paramount importance.
Two seemingly slightly different forms of idiopathic thrombocytopenic purpura exist. Classification into the acute and chronic forms is based on a somewhat arbitrary division by using 6 months' duration as the dividing line. The acute form of idiopathic thrombocytopenic purpura is more prevalent in children, usually following exanthems. The acute form has a stormier course but usually resolves spontaneously. The chronic form of idiopathic thrombocytopenic purpura is usually observed in young adults. The acute form affects both sexes equally, whereas the chronic form appears to affect females more often than males, with a female-to-male ratio of 2-3:1.
Idiopathic thrombocytopenic purpura mostly occurs with an onset of petechiae and/or ecchymoses on the skin and mucous membranes. In general, patients do not bleed, unless the platelets are additionally affected by the medications diminishing their function. If bleeding happens, its severity is correlated with platelet counts. Thrombocytopenia with 10 X 109 cells/L (10-50 X 103 cells/µL) can lead to spontaneous hemorrhages, petechiae and ecchymoses, and gingival bleeding and epistaxes, and GI or genitourinary bleeding. Intracranial bleeding is the most serious complication, but it is rare (< 1%). Idiopathic thrombocytopenic purpura responds poorly to platelet transfusion, with small or absent increments in platelet counts; however, in a patient who is bleeding, multiple platelet transfusions are still recommended, along with other treatment measures.
The goal of treatment in idiopathic thrombocytopenic purpura is to elevate the platelet count to a level adequate to prevent major bleeding. The standard of practice is to treat the patient with prednisone at 1 mg/kg given orally or with methylprednisolone sodium succinate (Solu-Medrol) given intravenously on a daily basis and tapered according to response in the platelet count. In 1 study, 39% of patients achieved complete remission, but only half maintained remission for more than 6 months after therapy was discontinued. [62] An alternative to oral prednisone therapy is high-dose oral dexamethasone 40 mg/d for 4 days. A single study of 125 patients showed an initial response in 85%, and 50% of initial responders did not require further treatment up to 5 years later. Of the patients who had a relapse, all responded to a second course of high-dose dexamethasone. [63]
In patients with refractory idiopathic thrombocytopenic purpura, high-dose immunoglobulins and immunosuppressive drugs have been tried with some success. IVIG and anti-Rh(D) both increase the platelet count within several days, with an effect lasting as long as several weeks. Additional therapies for chronic refractory idiopathic thrombocytopenic purpura include rituximab, cyclophosphamide, azathioprine, vinca alkaloids. Research is ongoing regarding the use of thrombopoietin for refractory idiopathic thrombocytopenic purpura.
Splenectomy is traditionally considered second-line therapy when initial steroid therapy fails to achieve a safe platelet count. With the availability of safe alternatives, such as rituximab and IVIG, splenectomy is now frequently reserved for third- or fourth-line therapy in patients without severe, life-threatening sequelae from idiopathic thrombocytopenic purpura.
Posttransfusion purpura
A sudden onset of mucosal bleeding and thrombocytopenia approximately 1 week after transfusion of RBCs characterizes posttransfusion purpura. Posttransfusion purpura most often affects multiparous women, but it can occur in anyone who previously received a transfusion. Posttransfusion purpura occurs because of autoantibodies produced against PL-A1 membrane–associated glycoprotein epitopes on host platelets.
The diagnosis of posttransfusion purpura is clinical; therefore, posttransfusion purpura should be considered in the differential diagnoses for any thrombocytopenia that occurs after a transfusion of blood products. Posttransfusion purpura may produce no cutaneous symptoms. [64] Posttransfusion purpura resolves within 3 weeks in most patients; however, because of its severity, treatment is required to shorten the duration. Steroids and high-dose intravenous immunoglobulins have been used successfully. The infusion of antigen-negative platelets may cause a febrile reaction, and they usually do not result in an increased platelet count. Plasmapheresis is usually successful but requires insertion of a large-bore catheter, which may be risky in these circumstances.
Disseminated intravascular coagulation and thrombotic thrombocytopenic purpura do not have much in common other than that they both are forms of nonimmunologic platelet destruction presenting as diffuse microangiopathy. This destruction causes fragmentation of RBCs, which can be readily observed on a blood smear review. Erythrocyte fragmentation and platelet destruction also occur in patients with stenosed or artificial heart valves and artificial vascular grafts. They all can cause purpuric lesions.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura is a fulminant, often lethal, disorder characterized by disseminated thrombotic occlusions of the microcirculation consisting of a pentad of Coombs test–negative hemolytic anemia, thrombocytopenia, neurologic symptoms (often focal deficits), fever, and renal dysfunction. Thrombotic thrombocytopenic purpura is more common in females than in males, with a female-to-male ratio of 2:1, and its peak incidence is in individuals aged 30-40 years.
Upon examination, multiple ecchymoses, jaundice, and pallor may be found. The clinical manifestations of thrombotic thrombocytopenic purpura result from consumptive thrombocytopenia and occlusion of the microvasculature, which, in turn, leads to organ dysfunction. RBCs are damaged while interacting with fibrin strands, producing schistocytes (fragmented RBCs) that are rapidly destroyed in the spleen.
Patients with thrombotic thrombocytopenic purpura rarely require consultation with a dermatologist, but skin biopsy or gingival biopsy is often requested. A confirmatory biopsy specimen demonstrates the hyaline microthrombi occluding the microvasculature; the microthrombi are composed of degranulated platelets and endothelial cell debris, periodic acid-Schiff–positive amorphous material, and variable amounts of fibrin. Both vascular inflammation and perivascular inflammation are absent. Biopsy specimens are diagnostic in only 50% of patients, and similar abnormalities are noted in association with disseminated intravascular coagulation, hemolytic uremic syndrome (HUS), and various forms of vasculitis. Hemolytic uremic syndrome may be regarded as a localized form of thrombotic thrombocytopenic purpura, in which the damage mostly affects the kidneys. The incidence of hemolytic uremic syndrome is higher in children, and, most of the time, it is preceded by an infection.
A deficiency of or an antibody directed against a specific von Willebrand factor (vWF)–cleaving protease leads to an accumulation of large vWF multimers, and platelet aggregation is thought to be responsible for some cases of thrombotic thrombocytopenic purpura. The current mainstay of treatment is plasma exchange with fresh-frozen plasma, which not only removes the antibody directed against vWF-cleaving protease but also supplies missing enzyme. Before its introduction, thrombotic thrombocytopenic purpura was associated with mortality rates greater than 80%. At present, the same percentage of patients survives the first episode of thrombotic thrombocytopenic purpura with aggressive plasmapheresis.
Corticosteroids, immunosuppressants, and rituximab have been used with some success in patients with primary refractory or relapsing thrombotic thrombocytopenic purpura. In 1 study, all 11 patients with refractory or relapsed thrombotic thrombocytopenic purpura and ADAMTS13 antibodies were treated with rituximab, and all had clinical remission. [65] Rituximab was well tolerated and may be a reasonable approach in patients with refractory or relapsed thrombotic thrombocytopenic purpura.
Disseminated intravascular coagulation
Disseminated intravascular coagulation has been documented to be associated with many disorders, including obstetric complications, infections, neoplasms, acute promyelocytic leukemia, antiphospholipid syndrome, and massive tissue injury. As many as 60% of patients with disseminated intravascular coagulation have skin lesions, which can be an initial manifestation. All forms of purpura can be observed, including hemorrhagic bullae and purpura fulminans, which can progress to peripheral gangrene. Disseminated intravascular coagulation, as a result of widespread coagulation activation, causes precipitous consumption of coagulation factors. Typical laboratory findings in decompensated disseminated intravascular coagulation include thrombocytopenia, decreased fibrinogen level, prolonged prothrombin time (PT) and aPTT, and elevated levels of fibrin degradation products. The most effective therapy for disseminated intravascular coagulation is treatment of the underlying disorder.
Vitamin K replacement and transfusion of platelets and depleted factors (with fresh-frozen plasma and cryoprecipitate) may be beneficial. Heparin use has been beneficial in chronic disseminated intravascular coagulation, with improvement of laboratory values. Heparin use in acute disseminated intravascular coagulation has some discouraging results. Although substantially increased bleeding has not been documented, clinicians remain reluctant to use anticoagulants in this setting.
Qualitative disorders of platelet function
Whenever purpura is accompanied by a platelet count in the reference range, a platelet function disorder, which is always accompanied by a prolonged bleeding time, should be considered. The primary hemostatic mechanism involves multiple steps of platelets interacting with each other and with injured vessels. Abnormalities of multiple involved steps are the underlying reasons for a number of platelet defects reflected in a prolonged bleeding time. Certain medications (eg, aspirin, beta-lactam antibiotics, nitrates) can induce some of the platelet defects.
Bernard-Soulier syndrome is an autosomal recessive disorder (extremely rare) with mild thrombocytopenia, giant platelets, defective adhesion, and bleeding out of proportion to the reduction in platelet numbers. The platelets are deficient in Ib receptor, which is a receptor for vWF.
Glanzmann thrombasthenia is a rare autosomal recessive disorder that results from the inability of platelets to aggregate in response to adenosine diphosphate (ADP) or other platelets. Quantitative and/or qualitative abnormalities of membrane complex glycoprotein IIb-IIIa make platelet-to-platelet interaction impossible. The only available treatment is transfusion of normal platelets for emergencies.
von Willebrand disease is the most common congenital platelet abnormality, with a prevalence of 3-4 cases per 100,000 population. von Willebrand disease is heterogeneous, with several genetic patterns of inheritance. The basic defect is an abnormality or deficiency of vWF, which is required for normal platelet adhesion. The factor also acts as a carrier of factor VIII in the plasma. Because of the complex function, von Willebrand deficiency is associated with a complex hemostatic deficiency affecting both primary hemostasis and later steps of coagulation.
To diagnose von Willebrand deficiency, platelet tests and coagulation tests, including measurements of aPTT, factor VIIIc activity, ristocetin cofactor activity, and vWF antigen, are required. Desmopressin (dDAVP) is commonly used to correct the hemostatic defect in von Willebrand disease by causing the release of endogenous vWF from the endothelium. Platelet transfusion can be used when the condition fails to respond to initial dDAVP therapy.
Aprotinin reduces bleeding complications in patients undergoing coronary artery bypass grafting (CABG) or intracranial surgery. Aprotinin is now only available via a limited-access protocol. Fergusson et al reported an increased risk for death compared with tranexamic acid or aminocaproic acid in high-risk cardiac surgery. [66]
Hermansky-Pudlak syndrome is an autosomal recessive form of oculocutaneous albinism associated with a platelet abnormality. Deficiency of platelet storage granules results in mild episodes of bleeding in various organs, including the skin and the mucous membranes. Most patients are Puerto Rican. Hypopigmentation and pigmented nevi can occur in these patients. They also easily develop freckles in sun-exposed areas.
Platelet transfusion can be used in patients who do not respond to initial dDAVP therapy. Aprotinin has been shown to reduce bleeding complications in patients undergoing CABG or intracranial surgery. The most recent innovation is the use of recombinant factor VIIa (rFVIIa), which is thought to have a local procoagulant effect at sites of vascular damage through the binding of rFVIIa to the surface of activated platelets. Recombinant factor VIIa is expensive and should be reserved for severe life-threatening bleeding unresponsive to traditional therapy.
Hereditary disorders of connective tissue (eg, Ehlers-Danlos disease, pseudoxanthoma elasticum), steroid-induced purpura, purpura associated with paraproteins (eg, myeloma, amyloidosis), and cryoglobulinemia are a few examples of purpura not associated with thrombocytopenia or any particular platelet function abnormality.
-
Warfarin (Coumadin)–induced skin necrosis on the lower abdomen.
-
Sweet syndrome.
-
Sweet syndrome.
-
Pyoderma gangrenosum.
-
Pyoderma gangrenosum.
-
Koilonychia.
-
Koilonychia.
-
Urticaria pigmentosa (Darier sign).
-
Urticaria pigmentosa.
-
Urticaria pigmentosa.
-
Vitiligo.
-
Mastocytosis.
-
Mastocytosis.








