Background
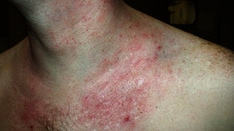
Epidermolytic ichthyosis (EI), formerly known as epidermolytic hyperkeratosis (EHK) or bullous congenital ichthyosiform erythroderma (bullous CIE), [1] is a form of congenital ichthyosis. It is inherited in an autosomal dominant fashion, with about 50% of cases representing spontaneous mutations. Epidermolytic ichthyosis presents at birth with erythroderma, blisters, and erosions and evolves over time into varying degrees of hyperkeratosis.
Pathophysiology
Epidermolytic ichthyosis results from heterozygous mutations in the genes encoding keratin 1 (KRT1) and keratin 10 (KRT10). [2] Mutations cause defects that compromise keratin alignment and assembly of intermediate filaments, leading to cellular collapse, blistering, and impaired barrier function. Compensatory hyperproliferation leads to hyperkeratosis.
Etiology of Epidermolytic Ichthyosis
Defects in genes for keratin 1 (KRT1) and 10 (KRT10) are the cause of epidermolytic ichthyosis. [3, 4, 5, 6, 7] Usually, these mutations are missense substitutions into the highly conserved alpha-helical rod and the nonhelical H1 domains of the keratin proteins. [8]
Palmoplantar keratoderma is usually associated with KRT1 mutations; however, in rare cases, palmoplantar keratoderma may be observed in patients with KRT10 mutations. [9] Novel mutations in both genes continue to be reported.
Patients with generalized epidermolytic ichthyosis may be born to parents with epidermolytic epidermal nevi (mosaic epidermolytic ichthyosis). [10, 11] Epidermal nevi with histologic changes of epidermolytic hyperkeratosis are caused by postzygotic mutations in keratin 1 or keratin 10. If the mutation also involves gonadal cells, which is thought to be more likely in patients with more extensive cutaneous involvement, affected individuals can have offspring with generalized epidermolytic ichthyosis.
Epidemiology
Frequency
The incidence of epidermolytic hyperkeratosis is estimated to be 1 in 200,000-300,000.
Race
No racial predilection is apparent for epidermolytic ichthyosis.
Sex
No sex predilection is recognized for epidermolytic ichthyosis.
Age
Epidermolytic ichthyosis is a lifelong condition with an onset at birth or in the neonatal period.
Prognosis
Epidermolytic ichthyosis is a lifelong condition. Some patients may experience amelioration of symptoms as they age. Risk for morbidity and mortality is highest in the neonatal period, where infants are at increased risk for complications such as sepsis and dehydration because of impaired barrier function. Later in life, affected patients may experience recurrent skin infections.
Patient Education
Educate patients with epidermolytic ichthyosis about the potential of passing the genetic defect on to offspring.
-
Pathology of epidermolytic ichthyosis (hematoxylin and eosin stain).
-
Pathology of epidermolytic ichthyosis (hematoxylin and eosin stain).
-
The scale in epidermolytic ichthyosis is classically described as "corrugated". Patients often experience erosions as a result of skin fragility.
-
The scale in epidermolytic ichthyosis is classically described as "corrugated". Patients often experience erosions as a result of skin fragility.
-
Palms and soles may have varying degrees of hyperkeratosis.
-
Hyperkeratosis involving the abdomen.
-
Hyperkeratosis involving the knee.