Overview
Palmoplantar keratodermas (PPKs) comprise a heterogeneous group of disorders characterized by persistent epidermal thickening of the palms and soles of the skin. Traditionally, PPKs have been categorized by their clinical phenotypes. As more is elucidated about the molecular genetics that contribute to these phenotypes, this classification scheme may change. Consequently, the authors of this review present each disorder by their traditional clinical divisions and offer updated genetic insights into each PPK entity.
The PPKs are classically divided into two subgroups based on whether they are inherited or acquired. Inherited PPKs are further divided into three distinct clinical phenotypes. The first inherited type is diffuse PPK, which describes uniform involvement of the palmoplantar surface. This pattern usually is evident within the first few months of life. The second is focal and striate PPK, which consists of localized areas of hyperkeratosis located primarily on pressure points and sites of recurrent friction. The third is punctate keratoderma, which features multiple small, hyperkeratotic papules, spicules, or nodules on the palms and soles. These tiny keratoses may involve the entire palmoplantar surface or may be restricted to certain locations (eg, palmar creases).
PPKs can be further classified based on the presence or absence of extracutaneous findings. In complex keratodermas, additional clinical features are associated with the PPK, such as lesions on nonvolar skin, abnormalities of adnexal structures, and/or abnormalities of other internal organs. Isolated PPK has none of these features.
Acquired keratodermas are divided into keratoderma climactericum, keratoderma associated with internal malignancy, PPK due to inflammatory and reactive dermatoses, PPK caused by infections, drug-related PPK, and systemic disease–associated PPK.
See the images below.
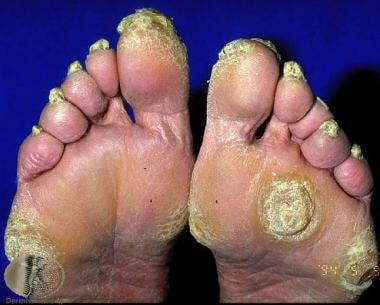 Focal palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/focal-kd2.jpg).
Focal palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/focal-kd2.jpg).
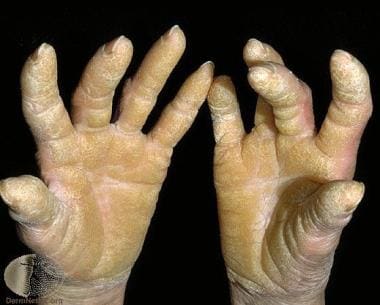 Diffuse palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/keratoder5.jpg).
Diffuse palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/keratoder5.jpg).
Diffuse Hereditary PPK
Diffuse types without associated features
Epidermolytic PPK (Vorner PPK)
Synonyms for epidermolytic PPK (EPPK) include diffuse Vorner disease and PPK cum degeneratione granulosa. In some ethnic groups, this form is the most common type of hereditary PPK. For example, it has an estimated prevalence of at least 4.4 cases per 100,000 population in Northern Ireland. It is inherited in an autosomal dominant fashion. Onset occurs in the first few months of life, but the disease is usually well developed by age 3-4 years.
Clinical features are very similar to diffuse nonepidermolytic PPK (NEPPK). A well-demarcated, thick, yellow hyperkeratosis is present over the palms and soles. An erythematous band is frequently present at the periphery of the keratosis. The surface is often uneven and verrucous. Painful fissures and hyperhidrosis are common. Finally, it is usually nontransgredient, with a sharp demarcation of the lesions at the wrists.
Histologically, keratinocytes show epidermolysis, hyperkeratosis, acanthosis, and papillomatosis. Perinuclear vacuolization and large keratohyalin granules are seen. Cellular breakdown in the spinous and granular cell layers occurs rarely and can lead to blister formation. Several biopsy specimens may be required to confirm the changes, as they may be subtle and patchy.
This disorder is most frequently associated with mutations in keratin 9, although keratin 1 has been implicated in a small number of reported cases. Mutations in keratin 9 typically result in phenotypes confined to the palms and soles. In contrast, keratin 1 is expressed ubiquitously, thus these mutations can affect the entire body surface. [1]
Topical therapies that have proven useful for EPPK include salicylic acid, keratolytic agents (ie, lactic acid and urea), and 50% propylene glycol in water under plastic occlusion several nights per week. Mechanical debridement with a blade also may be useful. Oral retinoid therapy has had variable effects and may not benefit patients with certain genotype profiles, such as K1 mutations. Finally, gene editing via CRISPR/Cas9, [2] as well as RNA-interference-based therapy, [3] has shown benefit in mouse models of this disease and may represent a powerful therapeutic strategy in the future.
Nonepidermolytic PPK (Unna-Thost PPK and Bothnian-type PPK [1]
Synonyms include diffuse Unna-Thost disease and PPK diffusa circumscripta. Diffuse NEPPK is inherited in an autosomal dominant fashion. The condition may manifest in the first few months of life but is usually well developed by age 3-4 years. It is another common type of hereditary PPK. Bothnian-type PPK has a prevalence rate of 0.3-0.55% in Northern Sweden (near the Gulf of Bothnia).
Clinically, waxy, thick, well-demarcated hyperkeratosis is present over the palms and soles. A red band is frequently present at the periphery of the keratosis. It is usually nontransgredient, with a sharp demarcation of the lesions at the wrists. Aberrant keratotic lesions may appear in the dorsum of the hands, feet, knees, and elbows. The dorsa of the fingers may be involved with a scleroderma-like thickening of the distal digit. A cobblestone hyperkeratosis of the knuckles may be seen. Nails may be thickened. Bothnian-type PPK may be distinguishable by the spongy-white appearance of affected areas when exposed to water.
EPPK and NEPPK show considerable clinical overlap and may be indistinguishable without histologic evaluation; however, some clinical features may help differentiate the two entities. NEPPK may have a more waxy, even appearance, compared with that of EPPK. Hyperhidrosis and pitted keratolysis may be present with NEPPK. Finally, secondary dermatophyte infections are more common in NEPPK.
Histologic findings include orthokeratotic hyperkeratosis associated with hypergranulosis or hypogranulosis and moderate acanthosis. Changes are nonspecific and common to many varieties of keratoderma. An absence of epidermolysis differentiates it from EPPK.
Unna-Thost PPK and Bothnian-type PPK are both autosomal dominant, but they differ in their associated mutations. Unna-Thost PPK molecular biology features include linkage to type II keratin locus on band 12q11-13, corresponding to a keratin 1 gene mutation. Bothnian-Type PPK is marked by a monoallelic missense mutation in the aquaporin 5 gene. Aquaporin 5 is expressed in eccrine sweat glands. [4]
Topical treatment options for NEPPK are similar to those for EPPK. These include salicylic acid, keratolytic agents (ie, lactic acid and urea), and 50% propylene glycol in water under plastic occlusion. Mechanical debridement with a blade may also be useful. Oral retinoid therapy has had variable effects. Treatment with an antifungal agent is beneficial if dermatophyte infection coexists with the NEPPK.
Mal de Meleda [5]
A synonym is keratosis extremitatum hereditaria trangrediens et progrediens. Mal de Meleda is an autosomal recessive disease. Onset occurs in early infancy, but the condition is rare. The prevalence is 1 case per 100,000 population. Initially, it was described in inhabitants of the Adriatic Island of Meleda (now called Mljet). [1]
Clinical features of mal de Meleda include a diffuse, thick keratoderma with prominent erythematous borders. Lesions are transgradient with spreading onto the dorsa of the hands and the feet. Constricting bands are present around the digits and can result in spontaneous amputation. Well-circumscribed psoriasislike plaques or lichenoid patches may be present on the knees and the elbows. Patients may have severe hyperhidrosis, possibly accompanied by malodor. Secondary bacterial and fungal infections are common. Perioral erythema; periorbital erythema and hyperkeratosis; nail changes (eg, koilonychia, subungual hyperkeratosis); and lingua plicata, syndactyly, hair on the palms and the soles, high-arched palate, and left-handedness are other clinical features.
Histologic findings include orthokeratosis, normogranulosis, and a pronounced stratum lucidum without epidermolysis. There is a prominent perivascular lymphohistiocytic infiltration.
Molecular biology features include mutations in the gene encoding SLURP-1 found on band 8q24.3. Proteins of the SLURP family have been implicated in transmembrane signal transduction, cell activation, and cell adhesion.
Treatment is with oral retinoids and topical keratolytic agents.
Nagashima-type PPK [6]
This condition is inherited in an autosomal recessive fashion. Onset of disease occurs between birth and age 3 years with stable disease severity over time. To date, the approximately 30 reported cases have occurred in Japan and China.
Clinically, the disease was initially described as a milder form of mal de Meleda. Some classify this keratosis as its own distinct entity, with common involvement of other sites, including the elbows and knees. Hyperhidrosis and tinea pedis infection are associated features. Case reports have described an increased prevalence of malignant melanoma in the hyperkeratotic lesions of Japanese patients with Nagashima-type PPK. [7] This may be due to the lack of epidermal Langerhans cells as seen on histopathology of hyperkeratotic skin. [8]
Treatment options include emollients and keratolytic agents.
Whole-exome sequencing has identified the molecular basis of Nagashima-type PPK as a defect in the SERPINB7 gene. The serpin superfamily of proteins is diverse and contributes to inflammation, immunology, and metastasis. [9]
Progressive PPK (Greither disease)
A synonym is transgrediens et progrediens PPK. This is inherited in an autosomal dominant fashion. Onset occurs in early infancy but may occur later in childhood.
Clinically, Greither disease is a transgradient PPK with extension of plaques onto the dorsa of the hands and the feet. There is characteristic involvement of the Achilles tendon. Scaly plaques may be found on the elbows, knees, and flexural areas. Hyperhidrosis and intrafamilial phenotypic variation are common. Pseudoainhum formation with amputation of the digits has been described.
Histologic features include epidermolysis of the granular cell layer. Lipid-laden corneocytes may be seen.
Molecular biology features include mutations in the gene encoding keratin 1. [10]
Treatment includes emollients, topical retinoids, keratolytics, and topical steroids.
Diffuse types with associated features
Mutilating PPK (Vohwinkel or Camisa syndrome)
Synonyms include PPK mutilans, loricrin keratoderma, and keratoderma hereditaria mutilans. Mutilating PPK is inherited in an autosomal dominant fashion. Onset occurs in infancy.
Clinically, this condition manifests in infants as a honeycomblike keratosis of the palms and the soles. It becomes transgredient during childhood. Later, constricting, fibrous bands appear on the digits and can lead to progressive strangulation and autoamputation. Starfish-shaped keratoses may occur on the knuckles of the fingers and toes, which is a characteristic feature of this disorder. Alopecia, hearing loss, spastic paraplegia, myopathy, ichthyosiform dermatosis, and nail abnormalities are associated findings. Cases of epithelioma cuniculatum have been reported.
Histologic findings include hyperkeratosis, acanthosis, and a thickened granular cell layer with retained nuclei in the stratum corneum.
Molecular biological studies have confirmed that the most common mutation found in Vohwinkel syndrome involves the gene GJB2, which encodes the gap junction protein connexin 26. [11] This subtype is associated with hearing loss. In contrast, a mutation in the gene for loricrin, involved in epidermal differentiation, is associated with mutilating keratoderma and ichthyosis, but not deafness.
Treatment includes oral retinoids. Reconstructive plastic surgery may be necessary for treatment of digital autoamputation. [12]
Also see Vohwinkel Syndrome.
Bart-Pumphrey syndrome [13]
A synonym is PPK with knuckle pads, leukonychia, and deafness. It is inherited in an autosomal dominant fashion. Onset occurs in infancy.
Clinically, all neonates are hearing impaired from birth and develop diffuse PPK in childhood. Leukonychia and hyperkeratoses over the joints of the hand also appear.
Molecular biological studies describe a new mutation in the GJB2 gene encoding connexin 26, which explains the clinical overlap with Vohwinkel syndrome.
Diffuse NEPPK and sensorineural deafness
This condition is inherited in an autosomal dominant fashion. [14]
Clinical features include diffuse palmoplantar hyperkeratosis in mid childhood preceded by slowly progressive, high-frequency hearing loss in early childhood.
Molecular biology features include a connexin 26 mutation. This mutation occurs on a distinct domain from that found in Vohwinkel syndrome. A mitochondrial point mutation has also been demonstrated as the cause of this phenotype, making this the only type of keratoderma associated with a mutation in mitochondrial DNA (serine tRNA). [11]
PPK with sclerodactyly (Huriez syndrome) [15]
PPK with sclerodactyly is inherited in an autosomal dominant fashion. Onset occurs in infancy.
Clinical features include red, atrophic skin on the dorsal hands and feet at birth. Diffuse, mild keratoderma is more marked on the palms than the soles. Other clinical features include sclerodactyly and nail abnormalities (hypoplasia, fissuring, ridging, koilonychia). PPK with sclerodactyly is also associated with marked atrophy and aggressive squamous cell carcinoma in areas of atrophic skin.
Histologic findings include acanthosis, accentuation of the granular layer, and orthokeratosis; Langerhans cells are almost completely absent in the affected skin. Under electron microscopy, dermoepidermal junctions and desmosomes are normal; however, dense bundles of tonofilaments are seen in the epidermal layer. The granular layer shows large, coarse, clumped keratohyalin.
Molecular biologic findings include a mutation in the gene mapped to 4q23.
Owing to the increased risk of skin cancer, close monitoring of patients is recommended. Other treatments include emollients, keratolytics, and topical and oral retinoids.
Hidrotic ectodermal dysplasia (Clouston syndrome)
This syndrome is an autosomal dominant disorder.
Clinical features include diffuse papillomatous PPK (especially over pressure points of the palms and soles), dystrophic nails, and hypotrichosis. Thickened, hyperpigmented skin may also appear over the small and large joints, including the knuckles, elbows, and knees. Thickened, severely dystrophic nails develop, but they may be normal at birth. Universal sparsity of hair affects the scalp, eyebrows, eyelashes, and axillary and genital regions. Sensorineural deafness, polydactyly, syndactyly, clubbing of fingers, intellectual disability, dwarfism, photophobia, and strabismus are associated features.
Clouston syndrome is mapped to 13q11. One form is caused by a mutation in the gene encoding connexin 30. Ultrastructural studies of the hair of these patients demonstrate disorganization of hair fibrils with loss of the cuticular cortex. Evidence reported in 2016 suggests that patients may be immunodeficient, with reduced phagocytic activity of granulocytes and monocytes. [16]
Mutilating PPK with periorificial keratotic plaques (Olmsted syndrome)
This type can be autosomal dominant, autosomal recessive, or X-linked recessive depending on the affected genes. Onset occurs in the first year of life.
Clinically, Olmsted syndrome begins focally in infancy and subsequently becomes diffuse. Later findings include flexion deformities and constriction of the digits, sometimes leading to spontaneous amputation. Progressive, well-defined perioral, perianal, and perineal hyperkeratotic plaques are present, as is onychodystrophy. Alopecia, deafness, nail dystrophy, and dental loss may be associated. Squamous cell carcinoma and malignant melanoma have developed in the areas of keratoderma. [17]
Histologic findings include hyperkeratosis without parakeratosis and mild acanthosis. Positive Ki-67 immunostaining of suprabasal keratinocytes suggests that hyperproliferation of the epidermis is a feature of this disease. [18]
Autosomal dominant and recessive forms have been associated with a gain-of-function mutation on the transient receptor potential vanilloid-3 (TRPV3) gene. X-linked recessive forms have been associated with mutations in the membrane-bound transcription factor protease, site 2 gene. [19]
Treatment includes oral and topical retinoids. Full-thickness excision and skin grafting has also been reported to result in clinical improvement. The development of a TRPV3 antagonist would provide the opportunity for targeted therapy.
PPK with periodontitis (Papillon-Lefèvre syndrome)
This condition is inherited in an autosomal recessive fashion. The prevalence of PPK with periodontitis is 4 cases per million. A variant, Haim-Munk syndrome, features, in addition to PPK and periodontitis, arachnodactyly, acroosteolysis, and onychogryphosis.
Clinically, diffuse transgredient PPK may be observed, typically developing within the first 3 years of life. Punctiform accentuation, particularly along the palmoplantar creases, may be seen. Unless treated, periodontosis results in severe gingivitis and loss of teeth by age 5 years. No significant correlation has been demonstrated between the level of periodontal infection and the severity of skin affections, which supports the concept that these major components of this syndrome are unrelated to each other. Patients exhibit increased susceptibility to cutaneous and systemic infections owing to neutrophil dysfunction. Scaly, psoriasiform lesions are often observed over the knees, elbows, and interphalangeal joints. Finally, patients may have malodorous hyperhidrosis. Reports from 2008 indicate a high prevalence of malignant melanoma in Japanese patients with Papillon-Lefèvre syndrome. [20]
Histologic findings include hyperkeratosis with irregular parakeratosis and moderate perivascular infiltration. Electron microscopic features include lipidlike vacuoles in corneocytes and granulocytes, a reduction in tonofilaments, and irregular keratohyalin granules.
Molecular biology findings include mutations in the CTSC gene. This gene codes for cathepsin C and mutations have been mapped to 11q14-q21. [21] Cathepsin C is a lysosomal protease known to activate enzymes that are vital to the body's defenses. Reports of multiple distinct mutations on the CTSC gene have been reported from consanguineous families in Turkey. [22]
Treatment includes oral retinoids for the PPK. Elective extraction of involved teeth may prevent excess bone resorption. Appropriate antibiotic therapy may be required for periodontitis and recurrent cutaneous and systemic infections. Early treatment with acitretin in childhood may allow patients to have normal adult dentition. Finally, in a 2018 in vitro study, introduction of recombinant cathepsin C partially restored some of the downstream immunologic functions of mutant cells and could represent an attractive therapeutic option in the future. [23]
Diffuse NEPPK with woolly hair and arrhythmogenic cardiomyopathy (Naxos disease) [24]
This condition is inherited in an autosomal recessive fashion.
Clinically, a diffuse, nontransgredient keratoderma with an erythematous border appears during the first year of life. Woolly (dense, rough, and bristly) scalp hair is present at birth. Cardiac disease, manifested by arrhythmias, heart failure, or sudden death, becomes evident during and after late puberty. Other cutaneous manifestations include acanthosis nigricans, xerosis, follicular hyperkeratosis over the zygoma, and hyperhidrosis.
Histologic findings include hyperkeratosis, hypergranulosis, and acanthosis.
Molecular biology findings include a mutation in the plakoglobin gene, mapping to 17q21, which is responsible for Naxos disease. Cardiomyopathy with alopecia and palmoplantar keratoderma (CAPK) is a subtype of Naxos disease described in a family with alopecia and right ventricular arrhythmogenic cardiomyopathy. CAPK has been linked to a mutation in the JUP gene coding for plakoglobin. [25] Plakoglobin is an important component of cell-to-cell and cell-to-matrix adhesion complexes of many tissues, including the skin and cardiac junctions. It also plays a role in signaling in the formation of desmosomal junctions. Mutations in the plakoglobin gene may lead to detachment of the cardiac myocytes, resulting in myocyte death. Plakoglobin mutations may also lead to desmosomal junction fragility in hair shafts, explaining the clinical phenotype of woolly hair.
Normalization of plakoglobin levels has been shown to restore cardiac function in mice and may be a viable therapeutic approach for improving the cardiac, and other, manifestations of this disease. [26]
Focal and Striate Hereditary PPK
Focal and striate PPK without associated features
Focal EPPK
Synonyms include keratosis palmoplantaris nummularis and hereditary painful callosities. This condition is inherited in an autosomal dominant manner. Onset occurs within the first 2 years of life. [27]
Clinical features include nummular keratotic lesions, mainly located on plantar pressure points. This condition is also painful. There is occasional blistering and minor nail changes.
Histologic features include local epidermolytic hyperkeratosis. [28, 29]
Molecular genetics have found associated mutations in the genes for keratin 6c and keratin 16. [1]
Striate PPK
Synonyms include Brünauer-Fuhs-Siemens syndrome, Wachter-type focal NEPPK, and PPK areata/striata. Striate PPK is inherited in an autosomal dominant fashion. Onset occurs in infancy or in the first few years of life.
Clinical features include marked variability in phenotypic expression. Hands may show minimal callous formation, or, changes may be undetectable in individuals with sedentary occupations. Friction-associated trauma from manual labor results in a striate pattern of PPK over the palmar aspects of the digits (islands of linear hyperkeratosis). Increased skin fragility may lead to skin splitting following trauma. Nails and hair may be involved.
Histology demonstrates acanthosis and an increased granular layer.
Molecular biology findings include mutations in desmosomal proteins, which have been implicated in this disorder. Desmosomal function is very important in areas prone to repeated friction. The three types of striate PPK are distinguishable by their molecular defects, as follows [30] :
-
Striate PPK I: Desmoglein 1 on 18q11-12
-
Striate PPK II: Desmoplakin gene on 6p21
-
Striate PPK III: Keratin 1
Treatment includes oral retinoids and topical keratolytics.
Focal and striate types with associated features
PPK associated with esophageal cancer (Howell-Evans syndrome)
Synonyms include tylosis esophageal cancer and focal NEPPK with carcinoma of the esophagus. This condition is inherited in an autosomal dominant fashion.
Clinically, focal PPK develops at age 5-10 years. The PPK is limited to the pressure points on the balls of the feet, with later mild involvement on the palms. Patients have an increased susceptibility to developing carcinoma of the esophagus (95% chance by age 65 y). Two variants have been described: one with a late onset of PPK and a higher risk of esophageal carcinoma, and another with an early onset and a benign course. Oral leukokeratosis and follicular keratosis are often present.
Histologic findings include acanthosis, a prominent granular cell layer, and hyperkeratosis of the palms and soles. Patients have thickened sweat ducts of the dermis, with the lumen often occluded by a hyperplastic epithelium.
The tylosis esophageal cancer gene (TOC) is localized to a small region on band 17q25, a region frequently deleted in persons with sporadic squamous cell esophageal tumors. The RHBDF2 gene in this region is involved in epidermal growth factor receptor (EGFR) signaling and also has a role in regulating keratinocyte proliferation. [31]
Key management includes surveillance for detection and treatment of esophageal dysplasia. This may include regular screenings with esophagogastroduodenoscopy and biopsies of suspicions lesions. Lifestyle and dietary modifications, such as smoking cessation and alcohol abstention, are important for risk minimization. The PPK can be treated with topical emollients, keratolytics, and oral retinoids. Genetic counseling may be offered to families once a diagnosis is established. [32]
Oculocutaneous tyrosinemia (Richner-Hanhart disease)
Synonyms include tyrosinemia type II. Oculocutaneous tyrosinemia is inherited in an autosomal recessive fashion.
Clinical features include focal, painful PPK development in childhood or adolescence. Occasionally, hyperkeratotic lesions develop on the elbows, knees, and tongue. Patients have hyperhidrosis and bullous lesions. Photophobia and herpetiform corneal erosions often appear in infancy and can lead to corneal ulcerations, scarring, and glaucoma. Tyrosine crystal deposits can be seen during slit-lamp examination. Intellectual disability develops, especially if patients are not treated.
Histologic findings include acanthosis with hyperkeratosis and hypergranulosis. Under electron microscopy, keratinocytes contain clumped tonofilament with adherent globoid keratohyalin granules and intracellular needle-shaped tyrosine crystalline inclusions can be found.
Molecular biology features include at least 15 mutations in the gene encoding tyrosine aminotransferase at band 16q22.1-q22.3. Deficiency of the enzyme tyrosine aminotransferase leads to increased levels of serum and urinary tyrosine and phenolic acid metabolites of tyrosine.
Treatment includes early institution of a low-phenylalanine and low-tyrosine diet; as such, newborn screening is essential. Additionally, supplementation of omega-3 has been shown to improve neurologic function in mouse models, although its role in mitigating the skin manifestations of the disease has not been studied. [33]
Pachyonychia congenita [34]
This condition is inherited in an autosomal dominant manner; however, a rare autosomal recessive pattern is also observed.
Clinical features include localized areas of hyperkeratosis on the palms and the soles, typically found on pressure points and weightbearing areas of the palms and soles. Patients have discoloration and thickening of the nails, which may be striking. Follicular hyperkeratosis, angular cheilitis, oral leukokeratosis, hyperhidrosis, painful blisters, and hoarseness may develop.
Type 1 is the Jadassohn-Lewandowsky type and is the most prevalent type of pachyonychia congenita. It is characterized by severe focal PPK, possibly with blistering. Type 2 is the Jackson-Lawler type. The focal PPK is milder in type 2 than in type 1. Natal or neonatal teeth, hair anomalies, and multiple pilosebaceous cysts (steatocystoma multiplex) are also present and help distinguish this type from type 1. Less common are types 3 and 4. Type 3 is the Schafer-Branuer type marked by corneal leukokeratosis in addition to the above clinical features. Type 4 is also known as pachyonychia congenita tarda and presents in the second or third decade of life.
Histologic features include hyperkeratosis with alternating orthokeratosis and parakeratosis. Large keratohyalin granules are present. Under electron microscopy, these granules appear as perinuclear keratin aggregates.
Molecular biology findings for the Jadassohn-Lewandowsky type include a keratin 6a gene mutation and keratin 16 gene mutations. The Jackson-Lawler type is associated with keratin 6b and keratin 17 gene mutations. Pachyonychia congenita tarda is associated with keratin 16 and 17 gene mutations.
Treatment includes emollients and keratolytics in mild cases and oral retinoids in more severe disease. Surgical excision of severely deformed nails offers a temporary solution, but nail dystrophy recurs. Injection of botulinum toxin has shown relief for hyperhidrosis and pain. A case report from 2018 suggests that topical sirolimus may be effective for reducing defective keratin production in the skin, leading to decreased plantar thickening and pain. [35] Finally, in a 2010 phase Ib trial, intralesional injection of short-interfering RNAs for the purpose of reducing mutant gene expression was shown to cause regression of plantar hyperkeratosis. [36]
Also see Pachyonychia Congenita.
Striate PPK with woolly hair and left-sided dilated cardiomyopathy (Carvajal-Huerta syndrome)
This condition is inherited in an autosomal recessive pattern. Both this disease and Naxos disease are autosomal recessive, whereas most of the hereditary dilated cardiomyopathies are autosomal dominant.
Clinical features include the appearance of striate PPK during early infancy. Left ventricular cardiomyopathy begins in adolescence and occasionally leads to early heart failure. Sudden cardiac death can occur in adolescence and early adulthood, making early cardiac management a priority. Woolly hair is present at birth. Striated lichenoid keratoses of the flexures, follicular keratoses on the elbows and knees, and clubbing of the nails have been described. Skin fragility, characterized by transient vesicles and blisters on the trunk and extremities, is another clinical feature.
Histologic features include spongiform edema with large intercellular spaces, clustering of desmosomes at infrequent sites of keratinocyte adhesion, and perinuclear localization of keratin in suprabasal keratinocytes, suggesting the presence of a collapsed intermediate filament network.
Molecular biology findings include homozygous mutations of the gene encoding desmoplakin, which maps to 6p24 and is the most abundant protein of the desmosome.
Severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome [37]
A synonym is PPK with erythroderma and hyperimmunoglobulin E. This condition is inherited in an autosomal recessive manner.
Clinical features include striate PPK at birth or during early infancy. Patients have erythroderma, ichthyosis, nail dystrophy, and diffuse hypertrichosis. Multiple food allergies, atopic dermatitis, high blood levels of immunoglobulin E, and eosinophilia are associated. Patients are at increased risk for bacterial skin infections, sepsis, and death at a young age.
Histologic features include acanthosis with hyperkeratosis and parakeratosis. A nonspecific dermal inflammatory infiltration may also be present.
Molecular biology has linked SAM syndrome to mutations in desmoglein 1 and, via whole-exome sequencing, to the N-terminal plakin domain of desmoplakin.
Punctate Hereditary PPK
Punctate PPK without associated features
Punctate keratosis of the palms and soles (Buschke-Fischer-Brauer type) [38]
Synonyms include punctate PPK type I, keratosis punctata palmaris et plantaris, Buschke-Fischer-Brauer disease, and keratosis papulosa. The prevalence is 1.17 cases per 100,000 population. This condition is inherited in an autosomal dominant manner, although sporadic cases have been reported. The age at onset is variable, between 10 and 70 years.
Clinically, asymptomatic, tiny, hyperkeratotic papules are present on the palmoplantar surface. Lesions are uncommon in childhood and usually manifest after age 20 years. This condition is not associated with hyperhidrosis. Patients commonly report pruritus. Most individuals lack associated features; however, spastic paralysis, ankylosing spondylitis, and facial sebaceous hyperplasia have been reported. An association with gastrointestinal and pulmonary malignancy is possible.
Histologic findings include substantial compact hyperkeratosis over a distinct area of epidermis, hypergranulosis, the presence of a cornoid lamella, and the absence of epidermal dyskeratosis or hydropic change, which help differentiate this condition from porokeratosis.
Linkage analyses have mapped the disease between bands 15q22.2 and 15q22.31. Monoallelic mutations in the AAGAB gene code for a protein with a GTPase domain involved in membrane trafficking and protein sorting. This syndrome is also associated with COL14A1 mutations.
Treatment includes keratolytics, topical salicylic acid, mechanical debridement, excision, and topical and systemic retinoids.
Punctate keratosis of the palmar creases
Punctate keratosis of the palmar creases occurs most commonly in African American patients aged 15-40 years. An autosomal dominant inheritance pattern has been suggested.
Clinical features include small, round depressions filled with conical keratinous plugs, which typically occur on the creases of the palms, fingers, and, less commonly, on the soles. The lesions are aggravated by friction, and, occasionally, they may be painful.
Histologic features include hyperkeratosis and parakeratosis.
Treatment may include keratolytics and topical retinoids.
Punctate porokeratosis of the palms and soles [30, 39]
Synonyms include punctate PPK type II, porokeratotic-type PPK, or spiny keratoderma. It is inherited in an autosomal dominant fashion.
Clinical features include tiny keratotic spines on the palms and soles beginning at puberty. Affected men have facial sebaceous hypoplasia.
Histologic features include columnar parakeratosis.
The molecular defect has yet to be identified.
Acrokeratoelastoidosis
Synonyms include punctate PPK type III, focal acral hyperkeratosis, or acrokeratoelastoidosis of Costa. Acrokeratoelastoidosis is usually sporadic, although familial cases suggest an autosomal dominant pattern of inheritance. Onset usually occurs before the second or third decade of life.
Clinical features include round or oval, shiny, firm, yellowish papules that can appear umbilicated. These papules are distributed along the marginal border of the palms, soles, and/or digits and can also be seen in the space between the thumb and forefinger, on the anterior surface of the lower legs, and over the knuckles and nail folds. The papules are usually asymptomatic.
Histologic features include focal hyperkeratosis and elastorrhexis (fragmentation and a decreased number of elastic fibers in the reticular dermis).
The causative mutation is unknown, although a case report from 2018 demonstrated a SLURP1 mutation in one patient with an acrokeratoelastoidosislike phenotype. [40]
No treatment is usually required for acrokeratoelastoidosis because lesions are asymptomatic. However, methods to remove the lesions for cosmetic purposes, including liquid nitrogen, salicylic acid, and topical tretinoin, have largely been unsuccessful.
Punctate types with associated features
Rare single-pedigree syndromes are as follows:
-
Punctate PPK with guttate hypopigmentation and calcinosis cutis (Cole disease)
-
Punctate PPK with cystic eyelids, hypotrichosis, and hypodontia (Schopf-Schulz-Passarge syndrome)
-
Punctate PPK with ankylosing spondylitis
-
Punctate PPK with facial sebaceous hyperplasia
-
Punctate PPK with spastic paralysis
-
Punctate PPK with lipomata
-
Punctate PPK with ichthyosis follicularis and bilateral severe sensorineural hearing loss [41]
-
Punctate PPK with congenital hip dysplasia [1]
-
Punctate PPK with nail dystrophy [1]
Acquired PPK
Keratoderma climactericum [42]
Synonyms include Haxthausen disease. Onset occurs in women of menopausal age.
Clinically, pressure areas on the soles are initially affected. Erythema and hyperkeratosis with fissuring make walking painful. Transgrediens is absent. Pruritus is minimal. Later in the course of the disease, palmar involvement is seen as discrete and centrally confined hyperkeratosis. Many patients are obese and hypertensive. A role for estrogen therapy has been proposed and is supported by the timing of onset and the response to topical and systemic estrogens. However, reports indicate that patients have normal hormone profiles. Some cases may represent a form of eczema or psoriasis.
Histologic features include compact orthokeratotic hyperkeratosis, hypergranulosis, irregular acanthosis with alternating thick and thin interpapillary ridges, and spongiosis with exocytosis of lymphocytes. In the dermis, a lymphocytic infiltrate is present around the upper dermal vessels.
Treatment includes topical estradiol 0.05% ointment or 25-40% urea.
Keratoderma associated with internal malignancy
Keratoderma is associated with malignancy as a paraneoplastic phenomenon and as a manifestation of malignancy predisposition. Skin changes tend to precede discovery of the neoplasm by a few months, and metastasis is usually present at diagnosis. Keratodermas have been associated with carcinomas of the esophagus, lung, breast, uterus, stomach, pancreas, kidney, bladder, colon, blood, and skin.
Acrokeratosis paraneoplastica of Bazex is primarily associated with squamous cell carcinoma of the aerodigestive tract, although other malignancies also have been implicated. [43] The condition affects white men aged 40 years and older. Psoriasiform changes of the fingers and the toes with nail dystrophy extend to involve the palms, soles, limbs, and trunk. The paraneoplastic nature of the condition is supported by a tendency for the severity of skin involvement to parallel the extent of tumor. The skin may clear with the removal of the tumor, but relapses occur with tumor regrowth and cervical metastasis. Also see Acrokeratosis Neoplastica.
Tripe palms (acanthosis palmaris) is a distinct cutaneous paraneoplastic syndrome most often associated with malignancy of the stomach or lung. The palms are diffusely thickened, with a velvety texture and ridged appearance. Malignant tripe palms persists, with less than one third of patients responding to tumor therapy. Tripe palms is also associated with bullous pemphigoid, psoriasis, and exfoliative dermatitis.
PPK due to inflammatory and reactive dermatoses
Chronic hand dermatitis is characterized by marked irritation, scaling, and fissuring. Causes may include atopic dermatitis, irritant contact dermatitis, or allergic contact dermatitis. Palmoplantar keratoderma in the setting of an eczematoid graft versus host disease has also been reported. [44]
Psoriasis may manifest as diffuse hyperkeratosis or as scaly plaques with a scalloped margin.
Keratoderma blennorrhagica occurs in 30% of patients with reactive arthritis. Lesions are compact, and the sole of the foot is the typical site of involvement.
Pityriasis rubra pilaris is associated with red-orange thick scale on the palms and soles with sharp borders. Usually, follicular papules with surrounding erythema are present, often on the dorsal proximal phalanges (see Pityriasis Rubra Pilaris).
Callosities develop in response to repeated trauma over bony prominences.
Lichen planus may cause an erythematous, scaly PPK or a punctate, yellow, warty keratosis. PPK and nail dystrophy have been seen in lichen nitidus.
Transient palmoplantar keratoderma has also been described as a marker of a distinct, severe subtype of treatment-resistant bullous pemphigoid. [45]
Aquagenic keratoderma [46]
Synonyms include acquired aquagenic PPK, transient reactive papulotranslucent acrokeratoderma, and aquagenic syringeal acrokeratoderma. Onset occurs during the second decade of life, and there is a predilection for adolescent females.
Clinically, within minutes of immersion in water, translucent whitish papules and plaques appear on the palms and, less commonly, on the soles. A burning sensation and edema of the hands may also occur simultaneously. The lesion resolves minutes to hours after drying of the affected skin. Hyperhidrosis may be associated with the condition. The pathogenesis of this condition is unknown but is thought to be related to increased skin water absorption (such as with defective skin barrier function) or possibly in association with increased sweat chloride concentrations (altered CFTR channel, hyperhidrosis). Other cases have been associated with the use of certain drugs, including cyclooxygenase inhibitors.
Histologically, the epidermis and dermis may be normal or mild orthohyperkeratosis, hypergranulosis, acanthosis, or dilated eccrine ostia may develop.
Treatment includes 20% aluminum chloride hexahydrate, which has shown significant clinical improvement in signs and symptoms. Other options include 12% ammonium lactate cream, 20% acid salicylic in petroleum jelly, 10% urea cream, iontophoresis, [11] and botulinum toxin injections.
PPK caused by infections
Dermatophytes manifest as hyperkeratosis affecting the soles, toe webs, and sides of the feet. Hyperhidrosis increases itching and malodor.
Human papillomavirus may form confluent exuberant masses on the palms and the soles, mimicking keratoderma.
Syphilis may manifest as a diffuse, symmetric keratoderma or papular PPK.
Leprosy can manifest as glove-and-stocking anesthesia, predisposing patients to infection, ulceration, and hyperkeratosis. Tuberculosis, especially miliary tuberculosis, may cause hyperkeratosis of the palms and soles.
Encrusted scabies may progress to strikingly hyperkeratotic and/or crusted lesions on the palms.
Patients with HIV infection may develop hyperkeratosis of the palms and soles as a manifestation of reactive arthritis, psoriasis, or syphilitic co-infection.
Drug-related PPK
Chronic arsenic exposure can lead to multiple, irregular, warty keratotic lesions on the palms and soles. Onset of the keratoses may occur 10-30 years after ingestion.
Drug hypersensitivity may result from agents such as glucan, verapamil, quinacrine, etodolac, mepacrine, proguanil, mexiletine, methyldopa, lithium, venlafaxine, gold, tegafur, fluorouracil, bleomycin, hydroxyurea, practolol, doxorubicin, dioxin, imatinib, vemurafenib, and capecitabine.
PPK may rarely occur as an adverse reaction to the influenza vaccine. [47]
Striate PPK has been linked to NRTI and NNRTI in case reports. [48]
Systemic disease–associated PPK
Myxedema (due to hypothyroidism) is associated with distinctive features that include its response to treatment and the severity and verrucosity of the hyperkeratosis, with diffuse involvement of the plantar surface and more limited involvement of the palms. The mechanism is unclear; however, it has been postulated that inhibition of carotene conversion to vitamin A by thyroxine deficiency may cause hyperkeratosis. Mild relief is achieved with thyroid hormone replacement.
Systemic lupus erythematosus has been linked to various morphologies of PPK and is an unusual skin manifestation of this autoimmune disorder.
Diabetes mellitus features include discrete plantar keratosis under the metatarsal arch and the great toe.
Chronic lymphedema and other circulatory disorders such as acrocyanosis and livedo reticularis have been associated with keratoderma.
Cutaneous T-cell lymphoma features diffuse hyperkeratosis accompanied by subungual hyperkeratosis and nail dystrophy, which is typical of Sézary syndrome.
Diets lacking protein energy and vitamin deficiency have been implicated in PPK with associated fissuring.
Idiopathic PPK
This is a diagnosis of exclusion.
Syndromes with PPK as an Associated Feature
These include the following:
-
Basal cell nevus syndrome
-
Bullous congenital ichthyosiform erythroderma
-
Cantu syndrome: Hyperkeratosis-hyperpigmentation syndrome
-
Cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma (CEDNIK) syndrome
-
Cole disease: Guttate hypopigmentation and punctate PPK
-
Congenital nonbullous ichthyosiform erythroderma
-
Darier disease
-
Dyskeratosis congenita
-
Ectodermal dysplasia with skin fragility
-
Epidermodysplasia verruciformis
-
Epidermolysis bullosa herpetiformis (Dowling-Meara type)
-
Epidermolysis bullosa simplex
-
Erythrokeratoderma variabilis
-
Familial pityriasis rubra pilaris
-
Hystrixlike ichthyosis-deafness (HID) syndrome
-
Ichthyosis hystrix of Curth-Macklin
-
Ichthyosis vulgaris
-
Incontinentia pigmenti
-
Keratitis, ichthyosis, and deafness (KID) syndrome
-
Kindler syndrome
-
Lamellar ichthyosis
-
Naegeli-Franceschetti-Jadassohn syndrome
-
Progressive symmetric erythrokeratoderma
-
Schöpf-Schulz-Passarge syndrome - PPK with hidrocystomas, hypodontia and hypotrichosis
-
Sjögren-Larsson syndrome
Treatment & Management
Treatment of all types of hereditary and nonhereditary keratodermas is difficult. An important hallmark of management involves screening for systemic illnesses, infections, culprit drugs (see list above), and neoplasia in patients with acquired PPK. Treating the underlying condition or stopping possible triggers is the most effective treatment. The most common therapeutic options only result in short-term improvement and are frequently compounded by unacceptable adverse effects. Treatment tends to be symptomatic and may vary from simple measures (eg, saltwater soaks, paring) to topical keratolytics, systemic retinoids, or reconstructive surgery with total excision of the hyperkeratotic skin followed by grafting. The mainstays of treatment are described below.
Topical keratolytics (eg, salicylic acid 5-10%, lactic acid 10%, propylene glycol 10-40%, urea 10-40%) are useful in patients with limited keratoderma.
Topical retinoids (eg, tretinoin) are effective, but treatment is often limited by skin irritation.
Topical calcipotriol ointment works by modulating epidermal differentiation and proliferation. It is sometimes used successfully to treat various forms of PPK, but randomized trials have not shown a significant therapeutic effect. [49]
Consider potent topical steroids with or without keratolytics in dermatoses with an inflammatory component.
Oral retinoids are effective, especially in some hereditary PPKs such as mal de Meleda, Papillon-Lefèvre syndrome, and erythrokeratoderma variabilis. Most hereditary PPKs require long-term treatment. Results indicate that acitretin is comparable to etretinate. Intermittent therapy should be attempted whenever possible. To limit unacceptable long-term adverse effects, the optimal dosage of acitretin in adults is 30-35 mg/d (0.5-1 mg/kg/d for adults and 0.5 mg/kg/d for children), with a maintenance dose of 25 mg/d. The optimal dosage of isotretinoin in adults is 1 mg/kg/d. Treatment of women of childbearing age results in long-term potential teratogenic effects. Caution is advised if the patient has an epidermolytic form because large erosions may occur with retinoid therapy. Some patients with KID syndrome have had worsening keratitis with retinoids. Patients should be started on a low dose, and the dose should be carefully increased to avoid flaring the disease and/or causing erosions.
Specific therapies dependent on the PPK type may be used. Psoralens and ultraviolet A (PUVA) or re-PUVA (a combination of oral retinoids and PUVA) may be indicated in persons with PPK secondary to psoriasis or eczema. Patients with oculocutaneous tyrosinemia may benefit from dietary restriction of phenylalanine and tyrosine. Improvement has been seen in persons with nonhereditary PPK after oral 1-alpha, 25-dihydroxyvitamin D-3. Symptomatic hyperhidrosis, a feature of many of the PPKs, can be treated with aluminum chloride. PPK caused or exacerbated by water may improve with iontophoresis or botulinum toxin. [11]
Careful selection of footwear and treatment of fungal infections are important.
Dermabrasion may permit increased penetration of topical agents, and carbon dioxide laser treatment may be beneficial in persons with limited keratodermas.
For severe and refractory keratoderma, consider surgery. Total excision of hyperkeratotic skin followed by grafting has been successful in a number of cases.
Paraneoplastic keratodermas are generally refractory to local treatment and may only respond upon successful treatment of the underlying neoplasm.
Many of the subtypes of PPK, including Huriez syndrome, Olmsted syndrome, Nagashima-type PPK, and Papillon-Lefèvre syndrome, are associated with an increased risk of skin cancer within hyperkeratotic lesions; as such, these patients should be closely monitored.
Novel future treatments for PPK may include gene-based therapies (gene editing, post-transcriptional gene silencing via short-interfering RNAs) and protein replacement therapy.
-
Diffuse palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/keratoder5.jpg).
-
Focal palmoplantar keratoderma. Courtesy of Professor Raimo Suhonen and DermNet New Zealand (https://www.dermnetnz.org/assets/Uploads/scaly/s/focal-kd2.jpg).




