Overview
The recognition and correct interpretation of cutaneous signs of diseases that primarily affect the bronchopulmonary system may aid the clinician in diagnosis and assessment of prognosis. This article reviews selected pulmonary diseases with distinctive cutaneous findings.
Cyanosis and Clubbing
Cyanosis
The distinctive blue or purple skin discoloration of cyanosis indicates the presence of at least 5 g/dL of desaturated hemoglobin. Severely anemic patients (regardless of the degree of desaturation) are, therefore, unable to manifest cyanosis. Cyanosis may be classified as central or peripheral, referring to the etiology of the hemoglobin desaturation, not to the observed anatomic location of the cyanosis. Central cyanosis is often a manifestation of congenital heart disease characterized by right-to-left pulmonary shunts. Central cyanosis typically involves the entire body but may be best appreciated on warm areas of the body (eg, tongue, oral mucosa, conjunctiva, skin). Peripheral cyanosis is a manifestation of diminished tissue perfusion with resultant increased local oxygen extraction leading to high levels of desaturated hemoglobin. Shock, heart failure, and peripheral vascular disease are common causes.
Clubbing
Clubbing describes the bulbous, clublike deformation of the distal portion of the fingers and toes resulting from connective-tissue proliferation (see the image below). The condition was first described by Hippocrates [1] ; an alternate term is Hippocratic fingers.
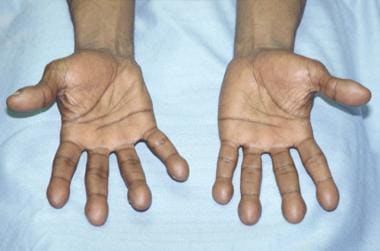 Clubbing. Photograph shows the palmar surface of the hands of a patient with marked clubbing. Note the bulbous clublike digits.
Clubbing. Photograph shows the palmar surface of the hands of a patient with marked clubbing. Note the bulbous clublike digits.
Clubbing may be hereditary (ie, primary hypertrophic osteoarthropathy, which is discussed in the next section), but, more often, it is acquired and is associated with a number of infectious, inflammatory, neoplastic, and vascular disorders. Although it may occur with several lung diseases (ie, lung cancer, tuberculosis, lung abscess, bronchiectasis, cystic fibrosis, idiopathic pulmonary fibrosis), [2] clubbing is a distinctly unusual finding in chronic obstructive pulmonary disease and its presence should prompt the clinician to search for other causes (particularly lung cancer). [3, 4]
Pathophysiology
The pathogenesis of clubbing has not been definitively determined. One promising hypothesis suggests that platelet-derived cytokines may play a central role. [5] This hypothesis proposes that diseases that disrupt the pulmonary circulation and allow right-to-left shunting of blood prevent the normal fragmentation of megakaryocytes in the pulmonary vasculature. If the megakaryocytes gain access to the systemic circulation, then lodge in the microvasculature of the digits, platelet-derived growth factor and/or vascular endothelial growth factor are released. [6, 7] These cytokines may induce a cascade of events (eg, increased vascular permeability, increased smooth muscle cell and fibroblast production, monocyte and neutrophil chemotaxis) that ultimately results in clubbing. A wide variety of malignant tumors, including lung cancer tumor, are also known to produce vascular endothelial growth factor.
Histologic findings may correspond to the duration and severity of disease. Early findings include dermal fibroplasia. Later, mucoid degeneration of the ground substances occurs. Severe long-standing clubbing is characterized by interstitial edema and infiltration by plasma cells and lymphocytes. Connective-tissue proliferation and interstitial edema result in a characteristic spongy quality to the proximal nail fold—as if the nail were floating in the nail bed.
Clinical findings
Clinical examination findings may correlate with the severity of clubbing. The earliest finding may be the Schamroth sign, first described by a South African physician when he developed clubbing secondary to infective endocarditis. [8] In healthy individuals, a rhombus is observed when the dorsal surfaces of the right and left second fingers are placed against each other with the distal interphalangeal (DIP) joints and distal aspect of the nails touching. An absence of this rhombus is consistent with clubbing. The accuracy of this sign has not been examined.
The phalangeal depth ratio (see the image below) appears to be the simplest, validated bedside test. [9] If the depth of the finger at the proximal nail fold equals or exceeds that of the DIP joint (a ratio greater than or equal to 1), clubbing may be diagnosed.
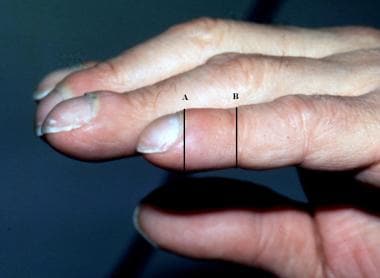 Clubbing, phalangeal depth ratio. Photograph shows clubbed fingers in profile. The phalangeal depth ratio refers to the ratio of the distal phalangeal to interphalangeal depth. Clubbing may be diagnosed when the distal phalangeal depth (A) is greater than the interphalangeal depth (B) (ie, phalangeal depth ratio >1).
Clubbing, phalangeal depth ratio. Photograph shows clubbed fingers in profile. The phalangeal depth ratio refers to the ratio of the distal phalangeal to interphalangeal depth. Clubbing may be diagnosed when the distal phalangeal depth (A) is greater than the interphalangeal depth (B) (ie, phalangeal depth ratio >1).
Clubbed fingers also exhibit changes in nail-fold angles. Lovibond first observed that in healthy individuals, the nail exits the nail bed at an angle of approximately 160°. [10] The Lovibond (also called profile) angle approaches 180° in persons with clubbing. [9] The hyponychial angle is formed by a line joining the dorsal surface of the DIP joint with the dorsal surface of the proximal nail fold and a line joining the dorsal surface of the proximal nail fold with the hyponychium (thickened skin under the distal end of the nail). [11] A normal hyponychial angle is approximately 180°; mean values of approximately 195° have been obtained in children with clubbing secondary to cystic fibrosis. [9]
Lung Cancer, Hypertrophic Osteoarthropathy, and Superior Vena Cava Syndrome
Lung cancer
Although breast and prostate cancer are more prevalent in women and men, respectively, lung cancer remains the leading cause of cancer death in both sexes. Cutaneous, subcutaneous, or superficial lymph node metastases are relatively uncommon events in the pathobiology of lung cancer. However, these metastases may be encountered in clinical practice because of the relatively high prevalence of lung cancer.
Some variability exists in the reported rates of cutaneous metastases in lung cancer. One study of 1000 patients with unresectable lung cancer reported that 12% had a skin metastasis; an additional 4.8% had superficial lymph node involvement. [12] Based on data from a large tumor registry, another study reported 2.6% of patients with metastatic lung cancer were noted to have a skin metastasis. [13] Most studies report cutaneous metastases in 2-8% of cases of lung cancer. Only in rare instances does a skin nodule lead to the diagnosis of lung cancer. [13, 14, 15]
The most common sites for cutaneous metastases are the chest and abdominal wall, neck, and scalp. The cutaneous nodules are usually solitary and fast growing and mobile, painless, and firm (sometimes described as "rock hard"). Superficial lymph nodes are also typically firm and may be fixed to the adjacent skin. They are most often supraclavicular (mimicking the Virchow node seen in gastrointestinal malignancy) or in the anterior cervical lymph node chains, ipsilateral to the primary tumor (see the image below). Adenocarcinomas appear to have the greatest propensity to metastasize to the skin. [12, 13, 16]
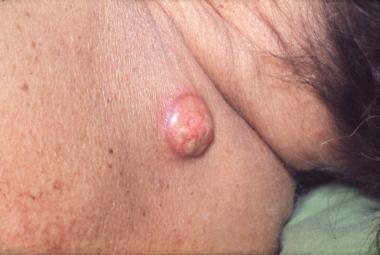 Lung cancer, skin metastasis. Photograph shows a skin metastasis on the upper portion of the back in a patient with lung cancer. Note the prominent superficial telangiectasia.
Lung cancer, skin metastasis. Photograph shows a skin metastasis on the upper portion of the back in a patient with lung cancer. Note the prominent superficial telangiectasia.
Importantly, a simple punch biopsy of a suggestive skin nodule may readily establish the diagnosis, as well as offering important prognostic information and obviating the need for additional staging procedures. Additionally, monitoring changes in the size of skin lesions is a simple way to monitor response to chemotherapy.
Hypertrophic osteoarthropathy
Clubbing associated with periostitis and arthritis is termed hypertrophic osteoarthropathy (HOA). HOA was described independently by E. von Bamberger in 1889 and Pierre Marie in 1890. HOA includes 2 distinctive clinical conditions.
Primary HOA
Primary HOA, also termed pachydermoperiostosis, is an autosomal dominant condition usually diagnosed in the second decade (typically around puberty), characterized by coarsening of the facial features as a result of sebaceous hyperplasia, cutis verticis gyrata, hyperhidrosis (especially of palms and soles), and severe seborrhea. [17] Arthritis is notably absent in persons with pachydermoperiostosis.
Secondary HOA
Secondary HOA is most frequently associated with lung cancer. [18] Symptoms of HOA commonly precede the diagnosis of underlying lung cancer by several months. [19, 16] HOA is more frequently associated with non–small cell lung cancer, particularly squamous cell carcinoma and adenocarcinoma. [20, 21, 22] Other conditions less frequently associated with secondary HOA include lung abscess, mesothelioma, primary biliary cirrhosis, ulcerative colitis, and Hodgkin disease.
Clinical findings
Patients often report bone or joint pain. Periostitis typically affects the distal portions of the long bones of the leg or forearm, and bony tenderness is found in these areas during the physical examination. [19, 23]
Patients who present with acute polyarthritis may initially be misdiagnosed with rheumatoid arthritis (RA). Similar to RA, the knees, ankles, and wrists are most often affected. In contrast to RA, the rheumatoid factor is rarely positive and synovial joint fluid is noninflammatory (ie, < 500 leukocytes/µL, with few neutrophils and a low protein concentration).
Plain radiographs of symptomatic bones reveal periostitis in persons with well-established disease. However, early in the disease course, plain radiograph findings may be normal (approximately 50% of bone is resorbed when changes become evident on plain radiographs). Because it relies on increased osteoblast activity, radionucleotide bone scanning is a more sensitive modality to detect periostitis in the early stages, but it lacks specificity.
Differential diagnoses
The differential diagnosis of HOA includes clubbing, pachydermoperiostosis, thyroid acropachy, and acromegaly. Thyroid acropachy is seen in approximately 1% of patients with Graves disease and is usually accompanied by exophthalmos and pretibial myxedema. Patients with thyroid acropachy also may have clubbing and periostitis, although the periostitis is usually limited to the hands and feet, and the joints are spared.
Acromegaly is a result of a growth hormone excess and results in enlargement of the hands and feet in all patients. Overgrowth of bone and cartilage results in thick, blunted digits (without clubbing). Arthritis may occur but is due to degeneration of joints; radiographs reveal increased soft tissue along with tufting of the distal phalanges.
Treatment
Symptoms of HOA may respond dramatically to treatment of the underlying neoplasm, particularly if curative resection is possible. Partial symptomatic relief of pain related to periostitis or arthritis may be achieved with aspirin or nonsteroidal anti-inflammatory drugs. [24]
Superior vena cava syndrome
The superior vena cava (SVC) arises from the confluence of the right and left brachiocephalic veins. It travels approximately 7 cm before emptying the venous drainage of the upper body (ie, head, neck, arms, chest) into the right atrium. SVC syndrome occurs when blood flow through the SVC is impeded, either by extrinsic compression or intraluminal narrowing.
A 1992 review underscores the importance of observing and interpreting the dermatologic findings in this condition. [25] Many patients with this syndrome were initially diagnosed by dermatologists after referral for evaluation of a rash on the upper chest. Malignant neoplasm is by far the most common cause. Thrombosis of the SVC is encountered more frequently in critically ill, hospitalized patients in association with intravascular devices (eg, central venous catheters, pulmonary artery catheters, pacemakers).
Etiology
Lung cancer is the most common cause of SVC syndrome. Because of the tendency to arise centrally (near the SVC), small cell carcinoma is the most common histology. Lymphomas, specifically the non-Hodgkin variety, are the next most common etiology. Other, much less common etiologies for SVC syndrome include mediastinal fibrosis, most commonly secondary to histoplasmosis, and retrosternal goiter.
Clinical findings
Markedly dilated veins or venules (often in vertical parallel clusters on the chest above the level of the heart) constitute the earliest cutaneous finding in SVC syndrome (see the image below). [25] These result from increased collateral flow through the subcutaneous vessels of the chest wall. The engorged vessels shrink or disappear with relief of the obstruction of blood flow. Other signs that may result from the venous congestion include hoarseness, facial edema, plethora of the head and neck, conjunctival suffusion, and proptosis.
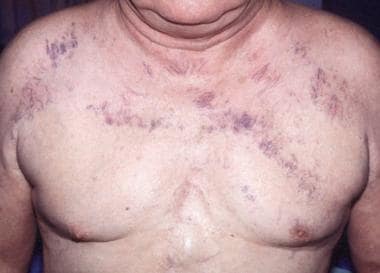 Superior vena cava syndrome in a 63-year-old man with known lung cancer who presented with dyspnea and facial swelling. The anterior chest is shown. Note the engorged, parallel, superficial venules. CT scanning of the chest confirmed compression of the superior vena cava.
Superior vena cava syndrome in a 63-year-old man with known lung cancer who presented with dyspnea and facial swelling. The anterior chest is shown. Note the engorged, parallel, superficial venules. CT scanning of the chest confirmed compression of the superior vena cava.
Treatment
Although malignancy-related SVC syndrome was previously viewed as an oncologic emergency that required immediate radiation treatment, even before tissue diagnosis, now recognized is that most patients do not require emergent radiotherapy. [26] In fact, radiotherapy prior to biopsy may make histologic diagnosis and subsequent treatment decisions difficult. [27] Exceptions include patients with life-threatening laryngeal or cerebral edema or tracheal compression resulting in respiratory compromise.
Initial conservative management, including elevation of the head of the bed and supplemental oxygen, often provides symptomatic improvement and allows sufficient time to obtain a tissue diagnosis. Diuretics and corticosteroids are often used, but their efficacy is not well established. Anticoagulants and thrombolytics may be used in the setting of intravascular thrombosis (but may cause intracranial hemorrhage in patients with malignancy-related SVC syndrome and brain metastases). Intraluminal stenting can also be considered for patients who require rapid symptomatic improvement.
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disease of unknown etiology. [28] Almost all patients have involvement of the respiratory tract at some point during the disease course. Patients often present with dyspnea, nonproductive cough, and pleuritic chest pain. Nonspecific symptoms, such as fatigue, malaise, anorexia, weight loss, and low-grade fevers, are also common. [29]
The disease is often first suspected when bilateral hilar adenopathy without parenchymal disease is discovered on a chest radiograph (see the image below), particularly in an asymptomatic patient. [30, 31]
 Sarcoidosis in a 20-year-old, asymptomatic woman. Marked bilateral hilar and paratracheal adenopathy are seen on this chest radiograph. Biopsy was performed and revealed noncaseating granulomas.
Sarcoidosis in a 20-year-old, asymptomatic woman. Marked bilateral hilar and paratracheal adenopathy are seen on this chest radiograph. Biopsy was performed and revealed noncaseating granulomas.
Chest radiographs may also show diffuse parenchymal disease with or without hilar adenopathy. The pattern of parenchymal disease is typically interstitial but also may be nodular. Pleural effusions are uncommon. Importantly, radiographic findings may not correlate with clinical findings. Pulmonary function testing typically reveals a restrictive pattern of disease with reduced lung volumes and decreased diffusion capacity.
Cutaneous involvement occurs in approximately a quarter to one third of patients but seldom is the sole manifestation of disease. [32] The presence of cutaneous sarcoidosis should prompt physicians to undertake a thorough evaluation. [33]
Cutaneous manifestations are described as either specific (histologically revealing typical noncaseating granulomas) or nonspecific. [34]
Etiology
Many theories have been proposed for the etiology of sarcoidosis, but none is universally accepted. Antigen exposure in genetically predisposed individuals is widely believed to be the common pathophysiologic pathway. Many believe that Mycobacterium subspecies are the causative agents, but definitive evidence is lacking. Although sarcoidosis may affect persons in any racial or ethnic group, it is more common in African Americans, Scandinavian populations, and Irish populations.
Pathology and diagnosis
The pathologic hallmark of the disease is the noncaseating granuloma. [28] Granulomas in the skin have the same histologic appearance as granulomas in the lung parenchyma. In patients with specific cutaneous manifestations, a skin biopsy may obviate the need for more invasive and expensive procedures. Other laboratory test results (eg, elevated serum angiotensin-converting enzyme levels, hypercalcemia, hypercalciuria) may be suggestive of sarcoidosis, but it is a diagnosis of exclusion.
Specific cutaneous manifestations
Maculopapular sarcoidosis is the most common specific cutaneous manifestation of sarcoidosis. [34] Lesions are red/brown-to-purple dermal papules and are typically seen on the face or extensor surfaces of the arms or legs. Papular sarcoidosis is usually associated with the acute forms of sarcoidosis and suggests a good overall prognosis. [35]
Lupus pernio is the most characteristic lesion of sarcoidosis and is often seen in older African American women. Lesions are violaceous, indurated plaques on the face and nose (see the image below) that resemble those seen in persons with chilblains (hence the name pernio). The cosmetic effects of lupus pernio may be particularly distressing to patients and may prompt them to seek medical attention. Unfortunately, patients with lupus pernio tend to have a more indolent disease course with a poorer overall prognosis than those with papular sarcoidosis. [35] Systemic disease manifestations, including pulmonary fibrosis specifically affecting the upper respiratory tract, chronic uveitis, and bone cysts, are also more common.
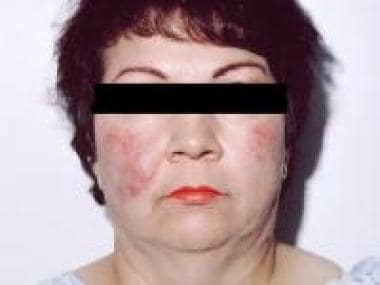 Lupus pernio in a patient with sarcoidosis. This cutaneous manifestation typically portends a more chronic disease course.
Lupus pernio in a patient with sarcoidosis. This cutaneous manifestation typically portends a more chronic disease course.
Subcutaneous nodules (Darier-Roussy sarcoid) may occur at any time during the disease course but most often are a manifestation of late-stage, chronic sarcoidosis. The subcutaneous nodules are firm, mobile, nontender, red/brown to purple, and 0.5-2 cm in diameter; they usually occur on the extremities and trunk and are often symmetrical. [36] They do not offer any clue to the prognosis. [35]
Cutaneous plaques may be round or oval, annular or arciform, and red/brown to purple (see the image below). The dermal infiltrates may be strikingly thick and tend to occur on the trunk and limbs. Plaques are a common manifestation of sarcoidosis and are associated with persistent pulmonary disease and more severe systemic involvement. [36, 35]
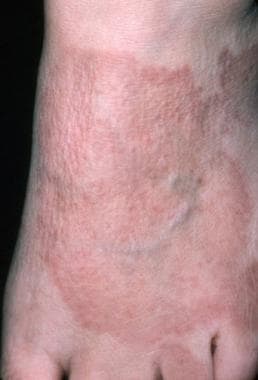 Sarcoidosis, cutaneous. The cutaneous manifestations of sarcoidosis are highly variable. The photograph shows an annular plaque on the dorsal surface of the foot. Biopsy revealed noncaseating granulomas typical of sarcoidosis.
Sarcoidosis, cutaneous. The cutaneous manifestations of sarcoidosis are highly variable. The photograph shows an annular plaque on the dorsal surface of the foot. Biopsy revealed noncaseating granulomas typical of sarcoidosis.
Preexisting well-healed scars from prior trauma or surgery may become red or purple and indurated as they are infiltrated with granulomas. Scar sarcoidosis can be confused with keloids. They may be seen in the acute phase, along with erythema nodosum (EN), but more often is associated with chronic disease. [35] Erythema nodosum is generally associated with a good prognosis. [36]
Nonspecific cutaneous manifestations
EN lesions are poorly circumscribed, tender, reddish nodules that resemble bruises and are located in the anterior tibial region. EN is associated with a number of conditions, mostly infectious. A 1998 retrospective study found sarcoidosis to be the second most common identifiable etiology for EN (streptococcal infections were the most common). [37] The lesions tend to erupt in crops and often occur in young white females. Sarcoidosis-related EN in combination with fever and uveitis is termed Lofgren syndrome. The prognosis of Lofgren syndrome is good; greater than 80% of patients experience spontaneous resolution of symptoms in less than 2 years. [35]
Extracutaneous manifestations
Cardiac involvement occurs in only 5-10% of patients but is associated with a poor prognosis. Most patients have rhythm or conduction disturbances noted on electrocardiograms. Anterior uveitis usually manifests as photophobia and blurred vision. Approximately 5% of patients have nervous system involvement; unilateral cranial nerve VII palsy is the most common manifestation. Liver infiltration is relatively common but is usually not clinically important. Liver function tests usually exhibit a cholestatic pattern (ie, elevated alkaline phosphatase levels and mild elevations of serum transaminase levels).
Treatment
Corticosteroids remain the mainstay of treatment for sarcoidosis and are generally reserved for patients with symptomatic pulmonary disease, hypercalcemia, uveitis, and neurologic or cardiac involvement. Severe, disfiguring cutaneous involvement may rarely require systemic corticosteroid therapy. Many other agents have been used in the treatment of cutaneous sarcoid (eg, topical corticosteroids, antimalarials, thalidomide, allopurinol, methotrexate) but none has been well studied. [35]
Wegener Granulomatosis, Churg-Strauss Syndrome, and Relapsing Polychondritis
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of unknown etiology that may affect any organ but most commonly involves the upper and lower respiratory tracts and the kidneys. Cutaneous manifestations are relatively common but usually not diagnostic.
Cutaneous findings
Histopathologic findings fall into 4 distinct categories: (1) necrotizing vasculitis, (2) granulomatous vasculitis, (3) lymphomatoid vasculitis, or (4) necrotizing palisading granuloma. [38, 39, 40] Skin findings are noted in approximately 10% of patients at presentation and develop in approximately 50% throughout the course of disease. [41, 42, 43] Purpuric lesions, specifically palpable purpura, are the most common and are usually found on the lower extremities. The second most frequent skin finding is necrotic ulcers, also typically found on the lower extremities. These lesions may be mistaken for pyoderma gangrenosum, but they lack the classic heaped-up border. Other cutaneous findings include subcutaneous nodules, papules, petechiae, and hemorrhagic bullae.
Up to 60% of patients may have oral mucosal disease; nonspecific oropharyngeal ulceration is most common. A rare and unusual gingivitis is considered pathognomonic of Wegener granulomatosis and is described as gingival hyperplasia with red petechiae and a friable granular appearance. Patients report pain and bleeding from the gums. Some evidence suggests that patients with cutaneous lesions have a higher incidence of renal involvement and that this may serve as an important prognostic indicator.
Clinical findings
The classic clinical triad of findings in Wegener granulomatosis is upper respiratory tract involvement, lower respiratory tract involvement, and renal disease. [41, 42, 43] Ear, nose, and throat involvement is present in more than 70% of patients at presentation, and lung involvement is present in approximately 50% of patients. Ear, nose, and throat findings include sinusitis, epistaxis, nasal septal perforation or saddle nose, or otitis.
Pulmonary disease may manifest as a nonproductive cough with or without hemoptysis and dyspnea. Less commonly, patients present with diffuse alveolar hemorrhage and renal failure; diagnostic confusion may arise in such cases (ie, Goodpasture syndrome vs Wegener granulomatosis). The finding of antineutrophil cytoplasmic antibodies (ANCAs) with diffuse cytoplasmic staining rather than antiglomerular basement (anti-GBM) antibodies is useful to distinguish these between the 2 entities.
Two types of ANCAs may be identified by immunofluorescence or enzyme immunoassay. [44] The diffuse cytoplasmic form of ANCA (c-ANCA) is characterized by granular staining with interlobular accentuation and is commonly detected in patients with Wegener granulomatosis. In most cases, the identified antigen is serine proteinase 3 (PR3). The test has a sensitivity of approximately 90% during active disease. Titers of cytoplasmic ANCA may correlate with disease activity (and may disappear in remission), but this correlation is not absolute. [44]
The perinuclear ANCA (p-ANCA) staining pattern is associated with antibodies that bind primarily to myeloperoxidase. The perinuclear ANCA pattern is less disease specific and may be seen in association with many different vasculitides, including polyarteritis nodosa (PAN), Churg-Strauss syndrome, microscopic polyarteritis, and Wegener granulomatosis.
The classic chest radiographic finding in Wegener granulomatosis is multiple, bilateral cavitary nodules, but less distinct parenchymal infiltrates or nodules are also commonly observed (see the image below).
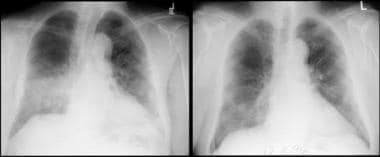 Wegener granulomatosis in a 71-year-old man. The left image shows right middle and lower lobe airspace disease. The patient was treated with steroids, and the hemoptysis resolved. The follow-up chest radiograph (right) shows radiographic improvement.
Wegener granulomatosis in a 71-year-old man. The left image shows right middle and lower lobe airspace disease. The patient was treated with steroids, and the hemoptysis resolved. The follow-up chest radiograph (right) shows radiographic improvement.
Bronchoscopy is frequently performed and may reveal tracheobronchial stenosis, focal or diffuse hemorrhage, or inflammatory or ulcerative lesions. Biopsy samples of these lesions usually show an inflammatory infiltrate but rarely reveal vasculitis. The presence of renal disease defines classic or generalized Wegener granulomatosis; the absence of renal involvement (ie, upper and/or lower involvement) is termed limited Wegener granulomatosis, but many of these patients eventually develop renal disease.
Treatment
Initial therapy often involves the combination of corticosteroids and cyclophosphamide and has dramatically improved long-term survival. [43] Untreated disease typically progresses rapidly and is nearly uniformly fatal (approximately 90% mortality rate at 2 y). Most patients can expect complete remission of their disease with treatment, although relapses are common.
Churg-Strauss syndrome
Churg-Strauss syndrome is a rare multisystem disorder that primarily affects the lungs, peripheral nervous system, and skin. Churg and Strauss' initial description of the syndrome in 1951 required histologic criteria to establish the diagnosis. Clinical and laboratory criteria have been more difficult to establish. Asthma, peripheral blood eosinophilia, and extrapulmonary vasculitis affecting 2 or more organs are cardinal clinical manifestations of Churg-Strauss syndrome. [45, 46]
The disease typically begins as allergic rhinitis in a patient without a prior atopic history. Mild asthma, a nearly invariable part of the clinical picture, then develops and progressively worsens. Cardiac disease, although less frequent than reactive airway disease, accounts for a substantial percentage of mortality. Cardiomyopathy results from endocardial and myocardial infiltration with granulomas or coronary arteritis.
Mononeuritis multiplex, similar to that seen with PAN, may be seen in as many as 70% of patients and usually involves the peroneal nerve with resultant foot drop. Another feature shared with PAN is gastrointestinal disease. As many as 60% of patients may have abdominal pain, which most often results from mesenteric ischemia secondary to mesenteric vasculitis.
Chest radiographs typically reveal characteristic bilateral, patchy, and nonsegmental fleeting infiltrates. [47] Other less common intrathoracic manifestations include pleural and pericardial effusions.
Laboratory findings include striking peripheral eosinophilia, usually more than 1.5 X 109 cells/L. Eosinophilia is also common in patients with asthma; however, the magnitude is greater in patients with the Churg-Strauss syndrome. Eosinophils are the dominant cell in pleural or pericardial effusions. Other laboratory findings may include a positive perinuclear ANCA serology and elevated immunoglobulin E levels.
Cutaneous findings
As many as 70% of patients with Churg-Strauss syndrome have cutaneous manifestations. [48] A variety of lesions may be seen, the most common of which are palpable purpura, subcutaneous nodules, urticarial rashes, and livedo reticularis.
Palpable purpura usually involves the lower extremities and is common but nonspecific. Biopsy of these lesions reveals leukocytoclastic vasculitis. The most distinctive lesions are tender subcutaneous nodules occurring on the limbs and scalp, or, less frequently, on the trunk. Biopsies of these lesions reveal an eosinophilic granuloma, characterized by a necrotic core surrounded by densely packed eosinophils.
Treatment
Most patients with Churg-Strauss syndrome respond well to high-dosage corticosteroids. For those who do not, alternate therapies include azathioprine, cyclophosphamide, and intravenous immunoglobulin (IVIG).
Relapsing polychondritis
Relapsing polychondritis is a rare multisystem disorder of unknown etiology. [49, 50] The disease is characterized by recurrent, and typically progressive, bouts of inflammation of cartilaginous structures. The diagnosis is based on clinical findings (ie, no diagnostic test is available). Relapsing polychondritis predominantly affects whites in the fifth and sixth decades of life. A number of associated conditions have been reported, including several systemic vasculitides (ie, Wegener granulomatosis, PAN, Churg-Strauss syndrome), collagen-vascular diseases (ie, RA, systemic lupus erythematosus, reactive arthritis [Reiter syndrome], scleroderma, Sjögren syndrome), hematologic diseases (ie, leukemia, lymphoma), thyroid disease, ulcerative colitis, primary biliary cirrhosis, and diabetes.
Clinical findings
External ear pain and inflammation is a frequent presentation and an almost universal finding at some point during the disease course. Patients are routinely misdiagnosed with an infectious process (ie, auricular cellulitis). Physical examination offers an important clue—because only the cartilaginous portion of the external ear is affected in persons with relapsing polychondritis, the ear lobe is spared (see the image below). Other cutaneous findings include leukocytoclastic vasculitis, EN, and erythema multiforme.
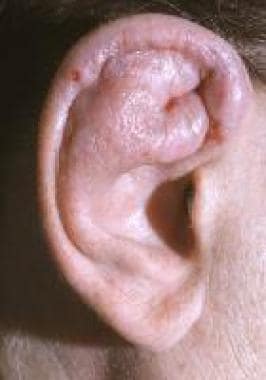 Patient with relapsing polychondritis and cartilaginous inflammation. Only portions of the external ear containing cartilage are affected, and the ear lobe is spared
Patient with relapsing polychondritis and cartilaginous inflammation. Only portions of the external ear containing cartilage are affected, and the ear lobe is spared
Most features of the disease share the common pathophysiologic pathway of cartilaginous inflammation. Saddle-nose deformity may occur as a result of destruction of the cartilage in the nasal septum (see the image below). A nonerosive nondeforming arthritis affecting both large and small peripheral joints occurs in as many as 85% of patients. Ocular inflammation is extremely common and may affect any part of the eye.
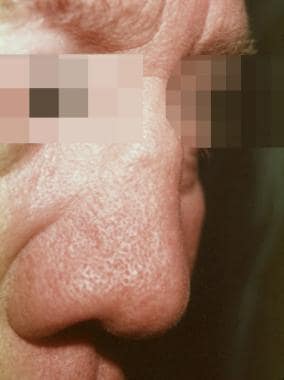 Relapsing polychondritis. Photograph shows saddle-nose deformity, which may be seen in persons with relapsing polychondritis as a result of the destruction of cartilage in the nasal septum.
Relapsing polychondritis. Photograph shows saddle-nose deformity, which may be seen in persons with relapsing polychondritis as a result of the destruction of cartilage in the nasal septum.
Destruction of cartilage in the laryngotracheal tree occurs in half to two thirds of patients and is the most feared disease manifestation. Respiratory distress results from destruction of the cartilaginous tracheal rings and subsequent tracheobronchial collapse or from inflammation of cartilaginous tissue and subsequent tracheal edema. Some patients may develop recurrent pneumonia because of insufficient clearance of secretions. Aortic root dilatation and subsequent aortic valve regurgitation is a less common, but potentially morbid, intrathoracic manifestation.
Treatment
A variety of immunosuppressive therapies have been used, but because of its rarity, clinical trial data are not available to guide treatment. In general, disease extent and severity are used to determine the intensity of treatment. Laryngeal, tracheal and bronchial involvement may result in stenosis or collapse and may require surgical intervention (resection and/or reconstruction) or placement of endotracheal or endobronchial tubes or stents. [51, 52]
Pulmonary Arteriovenous Malformations and Alpha-1 Antitrypsin Deficiency
Pulmonary arteriovenous malformations
Pulmonary arteriovenous malformations (PAVMs) are abnormal communications between pulmonary arteries and pulmonary veins. Most PAVMs are congenital. Approximately 70% of patients with a PAVM have hereditary hemorrhagic telangiectasia (HHT), also termed Osler-Weber-Rendu syndrome; conversely, 15-35% of persons with HHT develop PAVM. [53] HHT is associated with a mutation in the endoglin (HHT type 1) or ALK1 (HHT type 2) gene; the phenotypic expression of these 2 mutations is indistinguishable. [54]
PAVMs are typically multiple, bilateral, and located in the lower lobes. HHT affects multiple organ systems, including the skin, mucosal membranes, lungs, brain, and gastrointestinal tract. Recurrent epistaxis is frequently the earliest symptom and occurs in nearly all patients. Other extrapulmonary manifestations include notoriously difficult to manage gastrointestinal hemorrhages from arteriovenous malformations and central nervous system complications (eg, stroke, brain abscess) caused by paradoxical embolization through the PAVM.
Clinical findings
Pulmonary symptoms of PAVM result from the right-to-left shunting of blood. Patients may report fatigue, dyspnea, and platypnea (dyspnea worsens while upright and improves when supine). Although less common, hemoptysis may be the first symptom to prompt a patient to seek medical attention.
Physical examination may reveal nonspecific signs (eg, clubbing, cyanosis, occasional pulmonary bruit). Orthodeoxia (ie, decreased oxygenation while upright and improved when supine) is the physical examination correlate of platypnea and is the result of gravitational redistribution of blood flow to the lung bases (thus through the lower lobe PAVM) while the patient is upright. [55] Chest radiographs may reveal oval or round, homogeneous, nodular lesions several millimeters to several centimeters in diameter in the lung bases.
A clinical diagnosis of HHT can be suggested by examination of the skin. Telangiectasias of the face, hands, feet, chest, lips, tongue, and oral, nasal, and conjunctival mucosa become apparent in the second and third decades (see the image below). Lesions typically range from one to several millimeters in diameter, but they may grow much larger. Three-dimensional reconstructions of serial sections of skin biopsy specimens have established these lesions to be arteriovenous malformations.
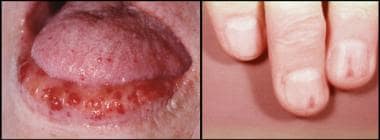 Hereditary hemorrhagic telangiectasia. Photographs show characteristic telangiectasias of the lips and tongue (left) and nail (right).
Hereditary hemorrhagic telangiectasia. Photographs show characteristic telangiectasias of the lips and tongue (left) and nail (right).
Treatment
The treatment of PAVMs consists of surgical resection or obliteration of the shunt by catheter embolization with coils (see the image below). A large or enlarging PAVM, pulmonary symptoms or signs (dyspnea or hypoxia), or paradoxical emboli are suggested indications for treatment. [54]
 Hereditary hemorrhagic telangiectasia in a 50-year-old man who presented with dyspnea. Workup revealed a large left lower lobe arteriovenous malformation (AVM). He was diagnosed with hereditary hemorrhagic telangiectasia. The pulmonary AVM was treated successfully using coil embolization. The image on the left shows an angiographic image of the AVM; on the right, the AVM is pictured after coil embolization.
Hereditary hemorrhagic telangiectasia in a 50-year-old man who presented with dyspnea. Workup revealed a large left lower lobe arteriovenous malformation (AVM). He was diagnosed with hereditary hemorrhagic telangiectasia. The pulmonary AVM was treated successfully using coil embolization. The image on the left shows an angiographic image of the AVM; on the right, the AVM is pictured after coil embolization.
Alpha-1 antitrypsin deficiency
Synthesized by hepatocytes, alpha-1 antitrypsin is the principal serum protease inhibitor. A single amino acid substitution results in a profound structural alteration of the enzyme, preventing its release from hepatocytes. Although perceived as a rare disease, the prevalence of alpha-1 antitrypsin deficiency may be as high as 1 case per 3000 persons in the United States. [56]
Clinical findings
The major organ systems affected by alpha-1 antitrypsin deficiency are the lungs, liver, and skin. The clinical manifestations are the result of unchecked proteinase activity that damages the parenchyma of these organs. Emphysema is the primary pulmonary manifestation of alpha-1 antitrypsin deficiency. Patients with alpha-1 antitrypsin deficiency develop severe fixed airflow obstruction at an early age, usually before age 50 years. Cigarette smoking greatly accelerates the progression of disease. Alpha-1 antitrypsin deficiency–associated emphysema occurs predominantly in the lung bases, as opposed to the upper lobe predominance seen in smoking-induced emphysema. Diagnostic testing includes quantitative (ie, determination of plasma alpha-1-antitrypsin levels) and qualitative (commonly referred to as phenotyping) studies.
Approximately 10% of children with the ZZ phenotype develop clinically significant liver disease; adults with cirrhosis of unknown etiology are occasionally found to express this phenotype.
Cutaneous manifestations
Recurrent ulcerative, necrotizing panniculitis is a rare extrapulmonary manifestation alpha-1 antitrypsin deficiency. [57, 58] The panniculitis is often preceded by trauma and is often mistaken for bacterial cellulitis. Crops of painful, red or violaceous, subcutaneous nodules occur on the trunk and proximal extremities. The nodules subsequently ulcerate and spontaneously drain a clear-to-yellow serosanguineous fluid, which may be oily as a result of liquefaction of fat.
Biopsy specimens reveal lymphohistiocytic infiltrate with areas of acute neutrophilic panniculitis. Culture results for bacteria, fungi, and mycobacteria are routinely negative.
Treatment
Intravenous infusions of purified alpha-1 antitrypsin, corticosteroids, dapsone, and doxycycline have been used to treat alpha-1 antitrypsin–related panniculitis.
Coronavirus Disease 2019 (COVID-19)
Multiple and extensive cutaneous symptoms have been reported in cases of coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Although COVID-19 largely manifests as a respiratory illness, dermatologic sequalae are common. One study estimates that 6% of all patients with COVID present with cutaneous symptoms. [59]
Clinical findings
Ghafoor et al found that maculopapular, morbilliform, and/or erythematous rash was the most common cutaneous symptom associated with COVID-19 infection. Urticaria and retiform purpura were the next most frequent signs. Skin infections and vesicular/pustular exanthem were also seen in a significant number of patients. [60]
The literature differs on the most common site for COVID skin manifestations. Some authors maintain the trunk is the most frequent location, while others assert that the acral regions of the lower extremities, specifically the feet and toes, are the most common site. Certainly, perniosis (chilblains) of the feet and toes (so-called "COVID toes") is a prevalent symptom in COVID infections. [61] This particular symptom has noted to be most common in pediatric patients and is associated with a milder clinical course. [62, 63]
Pathophysiology
Cytokine storm, including an inflammatory response to the spike (S) protein of SARS-CoV-2, vitamin D deficiency, angiotensin-converting enzyme 2 (ACE2) receptor activation, androgen levels, and psychological stress have all been proposed as mechanisms for the cutaneous sequalae present in many COVID infections. [64]
In SARS-CoV-2 infection, a type II transmembrane serine protease (TMPRSS2) enzyme activates the ACE2 receptor subsequent to the attachment of the S protein to ACE2. This binding and TMPRSS2 activation results in S protein activation and allows viral entry into the host cell through receptor-mediated endocytosis. Androgen receptors both promote and increase TMPRSS2 gene expression, and there is no other known regulatory mechanism for TMPRSS2. This relationship explains the less symptomatic course of COVID often seen in children who would have low relative expression of androgen receptors. [64]
Fat Embolism Syndrome
Fat embolism syndrome is a systemic disease that follows trauma and fractures of the long bones or pelvis. Fat embolism (without systemic manifestations) may also occur in association with hemoglobinopathies (sickle cell anemia), pancreatitis, bone marrow transplantation, and sepsis, and it may occur as a surgical complication of total hip and knee arthroplasty.
Clinical findings
The classic triad of findings in persons with fat embolism syndrome includes respiratory distress, altered mental status, and thrombocytopenia, typically occurring within 24-72 hours of long bone or pelvic fracture. The classic cutaneous manifestation (occurring in approximately one third to one half of patients) is an evanescent petechial eruption on nondependent portions of the body (ie, upper chest, neck, axillae, conjunctivae). [65] The finding is frequently overlooked but may be a key to diagnosis; it resolves in 5-7 days. Subsequent respiratory decompensation may range from mild hypoxemia to respiratory failure requiring intubation. Chest radiographs frequently show progressive, diffuse bilateral interstitial and alveolar infiltrates that are described as a "snowstorm pattern." Neurologic manifestations vary widely but typically are reversible upon resolution of the respiratory component.
Pathophysiology
The pathophysiology of respiratory compromise is uncertain, but theories include direct mechanical occlusion of the pulmonary capillaries by fat droplets released from the bone marrow or from the biochemical toxicity of these droplets once they are hydrolyzed to free fatty acids by lipases. [66]
Distinguishing between fat embolism (the presence of fat globules in peripheral circulation) and fat embolism syndrome (a systemic disorder that may be life threatening) is critical. Fat embolism occurs in as many as 90% of patients with long bone fractures; fat embolism syndrome is much less common (estimated rate of 1-2%).
Treatment
The mortality rate for patients with fat embolism syndrome is substantial (approximately 5-10%), and therapy is primarily supportive. Prognosis is primarily dependent upon the degree of respiratory compromise.
Yellow Nail Syndrome
First described in 1964 by Samman and White, yellow nail syndrome consists of the triad of yellow slow-growing nails, lymphedema, and pleural effusions. [67] The pathogenesis is unknown, but Samman and White postulated lymphatic impairment as the cause. One study suggests that a functional, rather than structural, lymphatic abnormality may be responsible. [68]
Clinical findings
Yellow (occasionally green) nail discoloration is the most obvious clue to diagnosis. The nails are characteristically thick and dystrophic with onycholysis, transverse ridging on an otherwise smooth surface, and loss of the cuticle. Women with yellow nail syndrome often color their nails with polish. The growth rate of fingernails is less than 0.5 mm/wk (approximately half the normal rate).
Lymphedema typically involves the lower extremities to a greater extent than the upper extremities and is symmetrical and nonpitting.
Pleural effusion is the classic pulmonary manifestation of yellow nail syndrome and usually the last in the triad to develop. The exudative effusions are characterized by a high concentration of protein (>3 g/dL) and lactate dehydrogenase (>200 IU/L), with fewer than 1000 leukocytes/µL, mostly lymphocytes. Effusions may be unilateral or bilateral and often recur after initial drainage. Other respiratory manifestations include bronchiectasis, recurrent pneumonias, bronchitis, and sinusitis.
Treatment
Treatment of pleural effusions may warrant pleurodesis (ie, instillation of a sclerosing agent into the pleural space), pleuroperitoneal shunts, or pleurectomy. [69]
Birt-Hogg-Dube Syndrome
Named after Arthur Birt, Georgina Hogg, and James Dubé in 1977, Birt-Hogg-Dubé syndrome is characterized by pneumothorax and lung cysts, benign skin tumors, and kidney tumors. [70]
It is a rare autosomal dominant condition caused by mutations in the FLCN gene, which normally provides instructions for making a protein called folliculin. Although the exact function is unknown, researchers believe that the FLCN gene is responsible for restraining cell growth and division. Hence, a mutation of the gene leads to the formation of tumors. However, the cause of the cyst and pneumothorax formation in the lung is not yet understood.
Clinical findings
According to the Birt-Hogg-Dubé foundation (BHD Foundation), every patient presents differently, even among members of the same family. The condition is characterized by skin lesions, renal tumors, and lung cysts or pneumothorax. It is estimated that 90% of patients have skin lesions, 90% have lung cysts, 25% have a collapsed lung, and 33% have kidney cancer.
Cutaneous manifestations
The most frequent skin features include fibrofolliculomas, trichodiscomas, and acrochordons. The cutaneous lesions are frequently on the face, neck, and upper torso and are described as multiple, pale-yellow, slightly raised, dome-shaped papules measuring 2-4 mm in diameter. [71]
Diagnosis and management
Diagnosis is suspected with the presence of multiple skin fibrofolliculomas, with at least one histologically confirmed. Differential diagnosis includes tuberous sclerosis. However, identification of the FLCN germline mutations is most supportive of the diagnosis. Even with this mutation, the manifestations can vary among all patients.
Other features include spontaneous pneumothorax, lung cyst(s), and renal tumors. The renal tumors are most frequently diagnosed at an early age, are bilateral, or are multifocal. They are often a hybrid pattern with chromophobe and oncocytoma features. These manifestations in a patient or the patient’s relatives can be diagnostic.
Testing for the mutation can lead to early diagnosis in family members. It is important to recognize the kidney tumors early and perform annual MRI scans of the kidney. Smoking cessation is recommended to prevention of pneumothorax. [72]
-
Clubbing. Photograph shows the palmar surface of the hands of a patient with marked clubbing. Note the bulbous clublike digits.
-
Clubbing, phalangeal depth ratio. Photograph shows clubbed fingers in profile. The phalangeal depth ratio refers to the ratio of the distal phalangeal to interphalangeal depth. Clubbing may be diagnosed when the distal phalangeal depth (A) is greater than the interphalangeal depth (B) (ie, phalangeal depth ratio >1).
-
Lung cancer, skin metastasis. Photograph shows a skin metastasis on the upper portion of the back in a patient with lung cancer. Note the prominent superficial telangiectasia.
-
Superior vena cava syndrome in a 63-year-old man with known lung cancer who presented with dyspnea and facial swelling. The anterior chest is shown. Note the engorged, parallel, superficial venules. CT scanning of the chest confirmed compression of the superior vena cava.
-
Sarcoidosis in a 20-year-old, asymptomatic woman. Marked bilateral hilar and paratracheal adenopathy are seen on this chest radiograph. Biopsy was performed and revealed noncaseating granulomas.
-
Sarcoidosis, cutaneous. The cutaneous manifestations of sarcoidosis are highly variable. The photograph shows an annular plaque on the dorsal surface of the foot. Biopsy revealed noncaseating granulomas typical of sarcoidosis.
-
Wegener granulomatosis in a 71-year-old man. The left image shows right middle and lower lobe airspace disease. The patient was treated with steroids, and the hemoptysis resolved. The follow-up chest radiograph (right) shows radiographic improvement.
-
Relapsing polychondritis. Photograph shows saddle-nose deformity, which may be seen in persons with relapsing polychondritis as a result of the destruction of cartilage in the nasal septum.
-
Hereditary hemorrhagic telangiectasia. Photographs show characteristic telangiectasias of the lips and tongue (left) and nail (right).
-
Hereditary hemorrhagic telangiectasia in a 50-year-old man who presented with dyspnea. Workup revealed a large left lower lobe arteriovenous malformation (AVM). He was diagnosed with hereditary hemorrhagic telangiectasia. The pulmonary AVM was treated successfully using coil embolization. The image on the left shows an angiographic image of the AVM; on the right, the AVM is pictured after coil embolization.
-
Lupus pernio in a patient with sarcoidosis. This cutaneous manifestation typically portends a more chronic disease course.
-
Patient with relapsing polychondritis and cartilaginous inflammation. Only portions of the external ear containing cartilage are affected, and the ear lobe is spared
Tables

What would you like to print?
- Overview
- Cyanosis and Clubbing
- Lung Cancer, Hypertrophic Osteoarthropathy, and Superior Vena Cava Syndrome
- Sarcoidosis
- Wegener Granulomatosis, Churg-Strauss Syndrome, and Relapsing Polychondritis
- Pulmonary Arteriovenous Malformations and Alpha-1 Antitrypsin Deficiency
- Coronavirus Disease 2019 (COVID-19)
- Fat Embolism Syndrome
- Yellow Nail Syndrome
- Birt-Hogg-Dube Syndrome
- Show All
- Media Gallery
- References








