Overview
Many disorders of the alimentary tract have dermatologic manifestations (see Table 1 below). A thorough understanding of the cutaneous/gastrointestinal (GI) relationship can alert the astute clinician to occult disease within the GI tract. This review attempts to explore this relationship by describing disorders involving both the GI tract and the skin. [1, 2]
Table 1. Dermatologic Manifestations of Alimentary Disorders (Open Table in a new window)
Dermatologic Manifestation |
GI Abnormality |
Disorder |
|
Periorificial granulomas [3] |
Malabsorption |
Crohn disease |
|
Koilonychia |
Esophageal webs |
Plummer-Vinson syndrome |
|
Liver disease |
Hemochromatosis |
||
Palmoplantar keratoderma |
Esophageal carcinoma |
Howel-Evans syndrome |
|
Acral rash |
Bazex syndrome [4] |
||
Telangiectasia [5] |
Esophagitis |
Scleroderma |
|
GI bleeding |
Hereditary hemorrhagic telangiectasia |
||
Cirrhosis |
Liver disease secondary to alcohol or other factors |
||
Vesicles/blisters/erosions |
Esophageal webs |
Epidermolysis bullosa |
|
Esophageal erosion |
Pemphigus vulgaris |
||
Pyloric atresia |
Junctional epidermolysis bullosa |
||
Hepatitis |
Porphyria cutanea tarda |
||
Malabsorption |
Dermatitis herpetiformis and celiac sprue |
||
Velvety hyperpigmented plaques, tripe palms, mucosal hyperplasia |
Gastric cancer |
Malignant acanthosis nigricans |
|
Yellowish papules, "chicken skin" |
GI bleeding |
Pseudoxanthoma elasticum |
|
Eruptive xanthomas |
Pancreatitis |
Primary biliary cirrhosis |
|
Biliary cirrhosis |
Sister Mary Joseph nodule |
||
Panniculitis |
Pancreatic |
Pancreatitis Pancreatic cancer |
Pancreatitis Pancreatic cancer |
Erythema nodosum |
Malabsorption |
Inflammatory bowel disease |
|
Skin tumors |
Multiple cysts |
GI polyps Colon cancer |
Gardner syndrome |
Sebaceous neoplasms, multiple keratoacanthomas |
Muir-Torre syndrome |
||
Trichilemmomas |
Cowden syndrome |
||
Hyperpigmentation |
Mucosal macular |
GI polyps Upper GI cancer |
Peutz-Jeghers syndrome |
Diffuse macular |
GI polyps GI cancer |
Cronkhite-Canada |
|
Diffuse |
Hepatomegaly Cirrhosis Hepatocellular carcinoma |
Hemochromatosis |
|
Plaques, acral |
Hepatitis |
Necrolytic acral erythema |
|
Lichenoid papules/plaques |
Hepatitis |
Lichen planus |
|
Hepatomegaly [6] Hepatic lesions |
Sarcoidosis |
||
Papules that heal with atrophic scars |
Perforation |
Malignant atrophic papulosis [7] |
|
Vascular malformation |
GI bleeding |
Blue rubber bleb nevus syndrome |
|
Ulcers with undermined borders |
Malabsorption |
Pyoderma gangrenosum and inflammatory bowel disease [8] |
|
Palpable purpura |
Abdominal pain |
Henoch-Schönlein purpura |
|
Plaques, intertriginous |
Malabsorption |
Acrodermatitis enteropathica |
|
Dermatology and the Pharynx
Kaposi sarcoma
Kaposi sarcoma (KS) is a neoplasm of vascular endothelial and lymphoreticular cells that can involve the skin and numerous visceral organs. The disease can manifest in four distinct forms: classic, iatrogenic, endemic, and epidemic. Originally described by Kaposi in 1872, the prevalence of the epidemic form of KS has risen dramatically in the United States over the past 20 years with the spread of AIDS, found in up to 30% of patients before the development of anti-HIV medications Further epidemiologic patterns, and the fact that it is more common in men who have sex with men compared with other groups at increased risk for HIV infection, suggested that KS is likely caused by an additional infectious agent. [9]
Human herpesvirus type 8 (HHV-8) has been implicated in the pathogenesis of KS in both HIV-infected and non–HIV–infected patients [10, 11, 12, 13] and is necessary but not sufficient for KS to develop. HHV-8 is contracted through contact with infected saliva, blood, or sexual activity and may lie latent for months to years. Tumors may develop as a result of HHV-8 in the setting of hyperglycemia, immune suppression, and viral co-infection. [14, 15] Evidence suggests that mTOR inhibitors like sirolimus (rapamycin) have the ability to prevent replication of HHV-8. [16]
The cutaneous lesions of KS (occurring primarily in the classic and epidemic forms) begin as pink or red macules, which darken and become increasingly papular as they enlarge. Although the classic KS lesions tend to occur on the legs and feet, the epidemic KS lesions are more common on the face and trunk.
Treatment of localized cutaneous disease includes radiotherapy, surgical excision, systemic therapy, or intralesional chemotherapy. In patients with widespread disease, careful consideration must be given to potential adverse effects. [17] Amongst the agents that attain a 30% response rate are interferons, antiviral agents (eg, zidovudine), cytotoxic agents (eg, vinblastine, etoposide), and combination therapy. For HIV-related KS with a low CD4 count or high viral load, immune reconstitution via antiretroviral therapy is the first-line therapy [18] ; other modalities include topical alitretinoin gel, daunorubicin, doxorubicin, paclitaxel, and interferon alfa-2b.
GI involvement occurs in 50-80% of patients with cutaneous KS and in almost 100% of those with oral lesions (see the image below). The GI tract may be involved at any level, although the most frequently afflicted sites (in order) are the small intestine, stomach, and esophagus. Oral lesions are most likely to affect the hard palate, followed in order of frequency by the gingiva and the tongue. In patients with non-HIV–associated KS, both the skin and GI lesions tend to have a relatively indolent course. In contrast, HIV-associated Kaposi lesions of the GI tract can have an aggressive clinical course, leading to extensive hemorrhage or partial small bowel obstruction. Treatment of symptomatic GI lesions involves laser ablation or surgical excision.
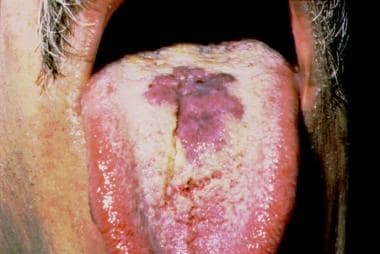 Oral Kaposi sarcoma in a patient with AIDS. Note the characteristic purple hemorrhagic papules coalescing into an irregular plaque.
Oral Kaposi sarcoma in a patient with AIDS. Note the characteristic purple hemorrhagic papules coalescing into an irregular plaque.
Dermatology and the Esophagus
Plummer-Vinson syndrome (Patterson-Brown-Kelly syndrome)
Signs of Plummer-Vinson syndrome include the mucocutaneous findings of koilonychia (brittle, spoon-shaped nails; see the image below) early loss of teeth, development of cheilosis, atrophy of the tongue, and angular stomatitis, along with iron deficiency anemia and the clinical complaint of dysphagia. [19, 20] The disease is rare in patients younger than 30 years, and women are affected in 80-90% of cases. Although the earlier diagnosis of iron deficiency and the ready availability of iron replacement have made this disease less common, the diagnosis of Plummer-Vinson syndrome nonetheless should be considered in patients presenting with long-standing intermittent dysphagia.
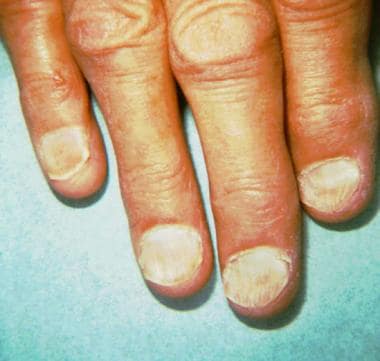 Koilonychia. Note the double concavity (longitudinal and transverse) of the nails.
Koilonychia. Note the double concavity (longitudinal and transverse) of the nails.
Complications include progressive painless dysphagia, which is described by patients as a choking sensation that worsens with solid foods. In 5-10% of patients with a postcricoid web, esophageal carcinoma eventually may develop (see the image below).
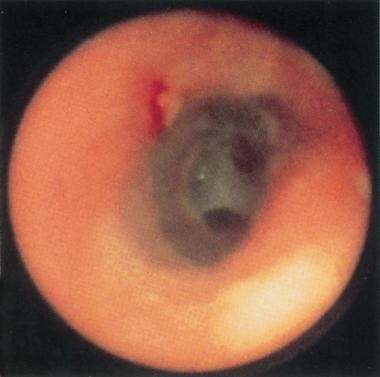 Postcricoid web in a patient with Plummer-Vinson syndrome. Note the 2 small openings within the web at the 2- and 6-o'clock positions, representing a significantly compromised proximal esophageal lumen. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
Postcricoid web in a patient with Plummer-Vinson syndrome. Note the 2 small openings within the web at the 2- and 6-o'clock positions, representing a significantly compromised proximal esophageal lumen. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
Diagnosis involves laboratory evaluation to confirm the presence of iron deficiency anemia and cineradiography to detect esophageal webs, which occur in the first 2-4 cm of the esophagus. Treatment involves iron supplementation, which often leads to rapid resolution of the dysphagia. In patients who continue to complain of dysphagia, therapy involves rupture of the esophageal web either by endoscopy or bougienage.
Epidermolysis bullosa
The name epidermolysis bullosa (EB) is given to a diverse group of rare inherited diseases that cause fragile skin. [21] The skin of these patients tends to form blisters at sites of minimal trauma, with symptomatology beginning at birth or early infancy. The disease is divided clinically into the following three forms depending on the layer in which blister formation occurs: dystrophic (beneath lamina densa), junctional (within the lamina lucida), and simplex (intraepidermal). Mutations in genes encoding structural proteins at the dermoepidermal junction and basement membrane have been implicated in different forms of EB. Many of these proteins are also expressed in a variety of different epithelia, including parts of the GI, urogenital, and respiratory tracts, leading to fragility and resulting in extracutaneous complication. [22]
A retrospective analysis of 223 patients with EB found 58% of patients had GI complications. [22] In EB simplex, constipation and gastroesophageal reflux (GOR) were frequently observed. In junctional EB, failure to thrive and protein-losing enteropathy (PLE) were the prominent findings. Constipation was common in patients with dystrophic EB. GOR and dysphagia affected most of the patients with recessive dystrophic EB, with two thirds also having significant esophageal strictures. Dysphagia most frequently presented in the first decade of life. The strictures seen are likely due to repetitive trauma from food ingestion. Lesions generally become symptomatic before age 30 years. Over half the patients with recessive-dystrophic EB required gastrostomy insertion. Diarrhea affected a small proportion of children with recessive dystrophic EB with changes of colitis in the majority of patients. In patients with severe EB of any type, nutritional deficiency, anemia, and growth retardation may develop over time, principally from the presence of oral and GI complications.
Systemic sclerosis (scleroderma)
Systemic sclerosis (SSc) describes a multisystem disease of unknown etiology that causes fibrotic changes in the skin, blood vessels, lungs, heart, kidneys, and GI tract. [23, 24] The disease affects all races and occurs worldwide, with clinical manifestations appearing most often in the third to fifth decade of life. SSc is divided into five forms: diffuse systemic sclerosis (dcSSc), limited systemic sclerosis (lcSSc), transitory form (dcSSc/lcSSc), systemic scleroderma sine scleroderma, and malignant scleroderma. The principal forms are dcSSc and lcSSc. Clinical manifestations of SSc are heterogeneous and vary as a result of type of disease (limited or diffuse) and organ involvement. In lcSSc (formerly called CREST [calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias] syndrome), skin tightening is confined to the fingers, hands, and forearms distal to the elbows, with or without tightening of skin of the feet and of the legs distal to the knees. In dcSSc, more rapid progression of skin fibrosis occurs, with proximal extremities and trunk involvement. Both dcSSc and lcSSc are associated with internal organ involvement; however, patients with dcSSc are at greater risk for clinically significant major organ dysfunction. Raynaud phenomenon is present in most patients with SSc and is often the earliest manifestation of disease. Skin changes usually begin with an early phase of skin edema, which presents as swollen fingers and hands, also known as the “puffy hand sign”.
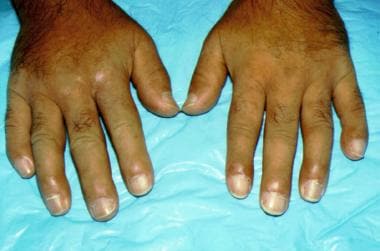 Scleroderma affecting the hands. Note the taut appearance of the skin and the curved nails.
Scleroderma affecting the hands. Note the taut appearance of the skin and the curved nails.
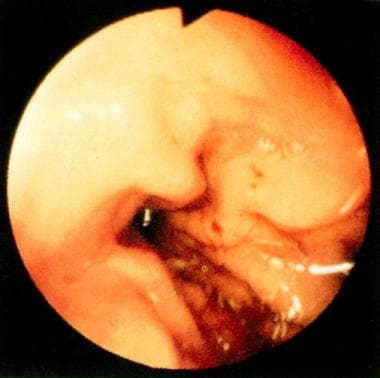 Telangiectases in the gastric mucosa of a patient with Osler-Weber-Rendu syndrome. The lesions can be seen most prominently at the 2-o'clock position proximally and the 3-o'clock position distally. Note the prominent red color of the lesions. Although these particular lesions appear flat, some GI telangiectasias may be slightly elevated. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
Telangiectases in the gastric mucosa of a patient with Osler-Weber-Rendu syndrome. The lesions can be seen most prominently at the 2-o'clock position proximally and the 3-o'clock position distally. Note the prominent red color of the lesions. Although these particular lesions appear flat, some GI telangiectasias may be slightly elevated. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
The GI tract is the most commonly involved internal organ in SSc. As many as 90% of patients with SSc demonstrate GI manifestations. [25] As in other organ systems, GI disease in SSc is thought to result from the excess collagen production, enhanced immunologic activity, and flawed cellular immune response, along with profibrotic growth factors like transforming growth factor-beta. The most commonly affected areas are the esophagus, the mid duodenum, jejunum, and large intestine.
Esophageal symptoms are the most common and include a feeling of premature fullness, reflux esophagitis, dysphagia, and epigastric pain, all of which stem from an incompetent lower esophageal sphincter and which are thought to occur in up to 90% of patients. As with other forms of reflux esophagitis, Barrett esophagus may develop, with the accompanying potential to transform to adenocarcinoma.
It is reported that stomach involvement occurs in 10-75% of SSc patients. [26] The gastric manifestations of SSc include gastric antral vascular ectasia (GAVE) and gastroparesis. GAVE can lead to chronic upper GI bleeding and iron deficiency anemia. Gastroparesis results is delayed gastric emptying or gastric paralysis, which results in early satiety, bloating, dyspepsia, nausea, and vomiting. [27]
Colonic involvement has been demonstrated by barium studies to affect between 10-50% of patients with scleroderma. Changes within the large intestine may result in chronic constipation, fecal impaction, and bowel obstruction. Involvement of the anal sphincter may lead to incontinence or even prolapse. Finally, overt or occult bleeding may develop as a result of trauma to telangiectasias, which may appear anywhere throughout the bowel. Treatment is symptomatic in all cases. Because the patient may require many years of therapy, careful consideration for the adverse effects of various promotility or antimotility agents should be undertaken.
The small intestine is the second most commonly involved portion of GI tract during SSc, following the esophagus. It is suspected that the small intestine is affected in 40% of SSc patients. Small intestine hypomotility is the primary abnormality, which can cause nausea, vomiting, bloating, distension, anorexia, and abdominal pain. Additionally, it can lead to pseudo-obstruction and bacterial overgrowth. Rarely, pneumatosis cystoides intestinalis (PCI), which is characterized by intramural gas in the GI tract, may occur. PCI generally has a good prognosis with no consequences; however, rarely it can cause intestinal ischemia requiring surgical intervention. [27]
Colon involvement is observed in 10-50% of SSc patients. Colon hypomotility is the most common complication, resulting in constipation and evacuation difficulty. Colonic telangiectasias are common and may cause overt bleeding, which can result in anemia. Anorectal involvement can also occur, leading to chronic diarrhea, fecal incontinence, and rectal prolapse. Neuropathy plays a role in the fecal incontinence. Liver and biliary involvement in SSc is relatively rare; however, primary biliary cirrhosis (PBC) is the most common hepatobiliary manifestation in SSc patients. The onset of PBC may precede, occur concomitantly with, or, more commonly, follow SSc onset. [27]
Treatment for SSc-related GI disease is aimed at both underlying disease management and symptom management. Treatments include proton pump inhibitors, H2 blockade, antacid tablets, and prokinetic agents for reflux and dysmotility symptoms. More recently, probiotics have also demonstrated efficacy in treating symptoms of reflux in patients with SSc. Probiotics can also aid patients with bacterial overgrowth, and fiber-rich foods can aid in managing constipation. Patients with dry mouth are prescribed artificial saliva, sugar-free gum, and candies. [28, 29]
Pemphigus vulgaris
Pemphigus vulgaris (PV) is a rare autoimmune blistering disorder primarily affecting the skin and mucosa. It is characterized by loss of cell-to-cell adhesion in the epithelial suprabasal layer, leading to superficial, easily ruptured blisters. Mucosal sites are commonly are involved, including the oral mucosa (often the presenting site), anus, cervix, and conjunctivae. Gastrointestinal involvement in particular is common throughout the GI tract, including GI bleeding, which can occur in up to 2% of patients with PV. [30] In 1970, Raque et al first described involvement of the esophagus, and it has been noted in multiple subsequent reports. [31, 32, 33] The prevalence of esophageal involvement in PV has not yet been firmly established, although a paper in The Lancet found evidence of esophageal involvement in 7 of 8 patients with PV who underwent endoscopy (87.5%). [32] Desmoglein 3, the target cell adhesion molecule for the PV autoantibodies, is strongly expressed in the esophageal epithelia. Symptoms of esophageal involvement include persistent sore throat, dysphagia, and odynophagia. In extreme cases, serious problems such as esophageal stenosis have been described. On endoscopy, the lesions can appear as bullae or irregular erosions covered in white exudate and an erythematous border. Sloughing of the entire esophageal lining (esophagitis dissecans superficialis) may also occur. Treatment for esophageal PV involves the administration of steroids or other immunosuppressive agents such as azathioprine, mycophenolate mofetil, methotrexate, or rituximab.
Acrokeratosis neoplastica (Bazex syndrome)
Acrokeratosis neoplastica is a psoriasiform (psoriasis-like) skin eruption, which most often occurs at the distal extremities, but can also involve the nose and helices of the ears. The disease progresses through three stages, spreading proximally. The first stage presents with poorly defined psoriasiform plaques involving the ears, nose, nails, fingers, and toes. Nail involvement is common in early disease stages, and the patient may report abnormal nails with painful nail folds. The second stage has more proximal involvement and a violaceous keratoderma with central clearing. The third stage is characterized by involvement of the legs, knees, thighs and arms.
Acrokeratosis neoplastica is highly associated with malignancy, particularly squamous cell carcinoma of the upper respiratory or upper digestive tract. [4] Notably, acrokeratosis neoplastica precedes the detection of malignancy in 65-70% of cases. [34] As such, when acrokeratosis neoplastica is suspected, a thorough history, focusing on cancer risk factors and symptoms, such as weight loss, malaise, and fatigue, should be performed to rule out malignancy. A thorough workup of the patient should also be done, with at least a chest radiograph, CBC count, and liver panel, but also other imaging directed by the patient’s symptoms.
Dermatology and the Stomach
Acanthosis nigricans
This relatively common skin finding appears as a brown-to-black, smooth, velvety plaque found in areas of skin folds, most commonly the neck, axillae, and groin (see the image below). An infrequent but important accompanying finding is the presence of acanthosis palmaris, or tripe palms, which appear as pronounced skin markings on the palmar surfaces of the hands together with diffuse velvety cobbled or honeycombed surface.
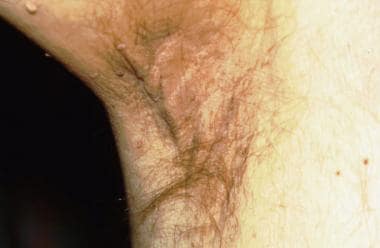 Acanthosis nigricans (AN) in a patient with pancreatic cancer. Note the papillomatous appearance of the axillary skin. This patient had previously been diagnosed with typical AN related to diabetes mellitus. After a long period of stability, the AN became much more severe and involved other parts of his skin, including the eyelids and scalp, prompting the search for malignancy.
Acanthosis nigricans (AN) in a patient with pancreatic cancer. Note the papillomatous appearance of the axillary skin. This patient had previously been diagnosed with typical AN related to diabetes mellitus. After a long period of stability, the AN became much more severe and involved other parts of his skin, including the eyelids and scalp, prompting the search for malignancy.
The mechanism responsible for the cutaneous lesions has not been identified. Tumor products may have their effect on skin through activation of insulinlike growth factors or their receptors in the skin. Alpha-transforming growth factors produced by tumor cells may have a role in malignant acanthosis nigricans through epidermal growth factor receptors found in the skin. One other hypothesis is that lytic factors produced by tumor cells may weaken the extracellular matrix of the skin, predisposing to acanthosis nigricans. Independent of the malignant variant, other identified associations include obesity, familial trait, diabetes mellitus, other endocrine disorders, or drugs (eg, corticosteroids, estrogens, nicotinic acid).
Of note, tripe palms can be diagnosed clinically by its distinctive appearance, and more than 40% of the time it is the first sign of an undiagnosed cancer. [35] Therefore, all patients with a diagnosis of tripe palms should receive a full workup for cancer, particularly lung or stomach cancer.
In patients with acanthosis nigricans, the possibility of an intra-abdominal malignancy should be considered, especially in patients without an obvious predisposing condition. [36] Clinical features such as a large number of plaques, weight loss, rapid progression of lesions, or the discovery of plaques in unusual locations, such as the lips, oral mucosa, hands, and genitalia, should be additional alerting factors. More than 90% of patients with tripe palms have an underlying malignancy, most commonly of gastric or pulmonary etiology. [35] The malignancies associated with acanthosis nigricans include adenocarcinoma (85% of cases), of which gastric carcinoma is present in 60%. Other reported sites of malignancy include the lung, bladder, endometrium, adrenal, bile duct, kidneys, thyroid, breast, liver, larynx, cervix, ovaries, prostate, and testicles. Nearly all cases are adenocarcinoma despite the site of origin. The skin lesions of malignant acanthosis nigricans often are discovered prior to the cancer diagnosis.
Prognosis is very poor for these patients, with a 1-year mortality rate of greater than 50%. In patients with successful tumor resection, the skin lesions often disappear spontaneously over time.
Sign of Leser-Trélat
The sign of Leser-Trélat is characterized by the appearance of multiple seborrheic keratosis (rough, well-demarcated, verrucous plaques with a “stuck-on” appearance), which rapidly grow in size and number. [37] It was originally named after Edmund Leser and Ulysses Trélat, but their reports actually described cherry angiomas rather than seborrheic keratosis. The sign of Leser-Trélat, in its currently accepted definition, was in fact described by E. Hollander. [38]
In addition to other cancers, these lesions are associated with GI adenocarcinomas, particularly of the stomach or colon. [39] The pathogenesis of the sign of Leser-Trélat in unknown, but it may be related to increased growth factor production by malignant cells. [40]
The sign of Leser-Trélat is controversial, as the incidence of seborrheic keratosis and malignancy both increase with age. [41] Therefore, it is particularly difficult to identify the sign of Leser-Trélat in the setting of an elderly patient. As such, this finding may be more useful in younger patients, when there is a rapid increase in the size and number of lesions or when other suspicious findings such as acanthosis nigricans, florid cutaneous papillomatosis, weight loss, dysphagia, or changes in bowel movements are present.
Similar to acanthosis nigricans, patients presenting with the sign of Leser–Trélat have a poor prognosis because the cancers associated with the sign of Leser-Trélat have poor outcomes. [42]
Florid cutaneous papillomatosis (Schwartz-Burgess syndrome)
First reported by Robert A. Schwartz and Gordon H. Burgess in 1978, florid cutaneous papillomatosis (FCP) is a rare disease. It presents with the rapid onset of numerous verrucous papules and nodules, which are very similar in appearance to viral warts. [43] Typically, these lesions first appear on the dorsal surface of the hands and wrists, and later spread to other body regions. FCP is most commonly associated with gastric adenocarcinoma but also can be associated with other malignancies. [44]
FCP can be difficult to distinguish from viral warts, and the diagnosis is most easily clarified by pathological evaluation of a lesion biopsy specimen. Clinically, FCP often occurs in conjunction with the sign of Leser-Trélat and acanthosis nigricans. Thus, it is important to look for these findings when FCP is suspected. [45]
Sister Mary Joseph nodule
As head nurse and skilled surgical assistant to William Mayo, MD, Sister Mary Joseph was the first to note that the presence of a periumbilical nodule heralded an advanced intra-abdominal malignancy (see the image below). [46, 47] The lesion was first coined "pants button umbilicus" by Dr. Mayo in 1928, and the Sister's name was not attached to the lesion until the 11th edition of Hamilton Bailey's Physical Signs in Clinical Surgery. The cutaneous finding is a firm, nontender nodule of red or purple hue that represents a metastasis from the primary tumor. The nodule most often results from contiguous extension of the tumor from the peritoneum, although lymphatic and venous pathways likely contribute in some cases. Approximately 90% of these malignancies are adenocarcinoma, with gastric or ovarian being the most commonly discovered primary malignancy. Other cited primary sites include the pancreas and bowel. The nodule is easy to biopsy and may save the patient from having to undergo diagnostic surgery. Of note, the biopsy aids in successfully diagnosing the primary malignancy site in only approximately half the cases.
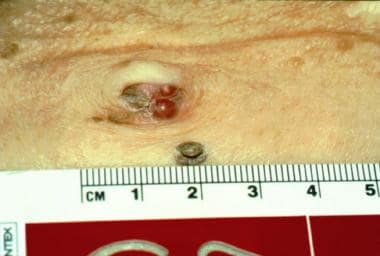 Sister Mary Joseph nodule in a patient with gastric carcinoma. Note the shiny, reddish, telangiectatic group of papules in the umbilicus.
Sister Mary Joseph nodule in a patient with gastric carcinoma. Note the shiny, reddish, telangiectatic group of papules in the umbilicus.
Osler-Weber-Rendu syndrome (hereditary hemorrhagic telangiectasia)
This autosomal dominant disorder occurs at a frequency of 1-2 cases per 10,000 population and is characterized by vascular dilatations of the skin as well as the oral, nasal, and GI mucosa (see the image below). [48, 49] First described by Babington in 1865 and further characterized by Rendu, Osler, and Weber, the diagnosis is based on the presence of three of the 4 following criteria: epistaxes, telangiectasia, visceral lesions, and an appropriate family history. [50]
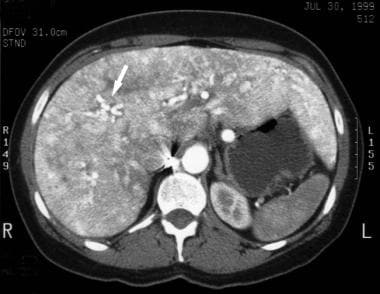 Arteriovenous malformations as seen on CT scan in a patient with Osler-Weber-Rendu syndrome. The patches of hyperdensity within the liver are the result of previous embolization procedures.
Arteriovenous malformations as seen on CT scan in a patient with Osler-Weber-Rendu syndrome. The patches of hyperdensity within the liver are the result of previous embolization procedures.
The dermatologic examination reveals numerous 1- to 3-mm macular or papular, sharply demarcated telangiectases on the face, lips, palate, tongue, ears, chest, or extremities, with occasional presentation under nails. The age of onset for the telangiectases is most often the third decade of life, although earlier presentations may occur during adolescence. Although the distribution of lesions and associated bleeding diathesis are clinically suggestive of hereditary hemorrhagic telangiectasia, it may occasionally be difficult to distinguish from similar cutaneous findings seen in generalized essential telangiectasia.
The most common and often cardinal clinical manifestation of Osler-Weber-Rendu disease is epistaxis, which generally starts in childhood and occurs in up to 80% of patients during their lifetime. The epistaxis results from spontaneous bleeding from telangiectases of the nasal mucosa and is generally recurrent. In about one half of patients, the frequency and seriousness of the epistaxis increases as the patient ages, leading to blood transfusions in as many as 30% of patients. Reported in 2019, treatments have included intralesional cyanoacrylate glue and bevacizumab. [51]
The second most common manifestation involves spontaneous hemorrhage from vascular telangiectasia within the GI tract. [52, 53] The GI lesions resemble the skin lesions in form and size and are most often found in the stomach or duodenum. A ring of less vascularized tissue surrounding the GI lesions is a characteristic finding on endoscopy. Bleeding from these telangiectases tends to begin in the fifth or sixth decade of life and often can be severe. Treatment modalities include endoscopic laser coagulation, bipolar electrocoagulation, surgical excision of severely affected bowel segments, or medical management with low-dose combination estrogen/progesterone or aminocaproic acid.
Another GI manifestation is the presence of arteriovenous malformations (AVMs), which can occur in the lungs, brain, and liver. Large AVMs can develop within the liver, leading to substantial shunt formation. Although the prevalence of these lesions is unknown, they have been blamed for portal hypertension with esophageal varices in some cases. Diagnosis involves the presence of elevated gamma-glutamyltransferase (GGT) or alkaline phosphatase levels, with confirmation by CT scan or color Doppler ultrasound. Treatment has tended to be conservative, with limited data on chemoembolization and surgical ligation suggesting that both may be effective when therapeutic intervention is deemed necessary.
Dermatology and the Liver and the Pancreas
Hemochromatosis
Hemochromatosis is a disorder of iron overload leading to excess deposition in multiple body organs. [54, 55] The hereditary form was first identified in the late 19th century as the classic triad of glycosuria (diabetes), bronze skin pigmentation, and cirrhosis. The dermatologic manifestation of the disease mainly involves skin hyperpigmentation. This discoloration has a characteristic metallic gray or bronze-brown color that is generally diffuse, but it may be increased in areas of scars or on the face, neck, extensor surfaces of the arms, and genitalia. Approximately 20% of patients also have pigmentation of the buccal mucosa or the conjunctiva.
Other characteristic skin findings in hemochromatosis include skin atrophy, ichthyosis, partial hair loss (most often in the pubic region), and koilonychia. [56] The characteristic skin changes of hemochromatosis are believed to result from increased melanin. The actual presence of iron seen in dermal sweat glands and the basal layer of the epidermis lead to the pigmentation seen in the late stages. Treatment of these patients with phlebotomy or chelating agents results in a decrease in skin pigmentation over time.
The most common GI finding in hemochromatosis is hepatomegaly, which is observed in more than 95% of symptomatic patients. Despite clinical findings of functional liver impairment (eg, hair loss, gynecomastia, testicular shrinkage), laboratory findings are generally unimpressive. Because hemochromatosis is not an inflammatory disease, liver enzyme (ie, aspartate aminotransferase, alanine aminotransferase) levels can be normal, even in advanced disease. In patients with long-standing untreated hemochromatosis, cirrhosis may develop, leading to hepatocellular carcinoma in as many as 30% of patients. Cirrhosis or its complications account for approximately 89% of hemochromatosis related deaths.
Overall, patients with hereditary hemochromatosis have an estimated 5% annual risk for hepatocellular carcinoma after the development of cirrhosis. Primary treatment involves phlebotomy and chelating agents, with conventional management of hepatic failure should this condition arise. Phlebotomy remains the treatment of choice according to 2019 American College of Gastroenterology Clinical Guidelines; however, emerging chelating agents hold promise as well. [57]
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common porphyria occurring in adults, with a prevalence ranging from 1 case in 5,000 to 1 case in 36,000 people. Porphyria cutanea tarda results from the decreased activity of the enzyme uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme biosynthetic pathway. Cutaneous findings are characterized by skin photosensitivity with increased skin fragility, facial hypertrichosis, blisters, scarring with milia formation, and skin hyperpigmentation on the hands and other sun-exposed areas. Porphyria cutanea tarda results from the decreased activity of the enzyme uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme biosynthetic pathway.
The disease can be either sporadic or familial, but, in either case, it is clear that extrinsic factors play an important role in producing a clinical manifestation of the disease. Endogenous and exogenous factors (eg, alcohol, iron, estrogen, porphyrins, chronic hepatitis C virus infection, polychlorinated biphenyls, polychlorinated cyclic hydrocarbons) produce oxidative stress on the liver, leading to inhibition or decreased production of UROD. The pathogenesis of iron in porphyria cutanea tarda also provides the link to hereditary hemochromatosis. All patients with porphyria cutanea tarda should undergo screening for hepatitis virus infection and hemochromatosis. [58, 59] Diagnosis of porphyria cutanea tarda involves the discovery of increased porphyrins in the blood, liver, stool, and urine.
The first step of treatment involves the removal of possible triggers, including iron supplementation, alcohol, and estrogens. If symptoms persist, repeated phlebotomy decreases the total iron load and leads to significant clinical improvement. Phlebotomy may induce clinical remission, reduce urinary porphyrins, and improve the sclerodermalike skin changes, but it is not proven to improve liver histology.
Other options include chloroquine and hydroxychloroquine, which form water-soluble complexes with the porphyrins, facilitating their excretion in the urine. Antimalarial therapy can be quite dangerous in patients with porphyria; it may precipitate massive hepatic injury. Therefore, doses should be initiated at low doses (125 mg, 2-3 times per wk) to reduce the likelihood of acute hepatic injury and retinopathy. Such therapy should be attempted only when other therapy has failed, with a clear understanding of the risks involved.
The prevalence of hepatitis C in patients with PCT has been measured in series of patients from many countries, with rates as high as 85% in these patients. [60, 61] Interestingly, multiple studies into the extrahepatic manifestations of patients with hepatitis C have failed to find an increased rate of PCT. This disparity may be explained by the fact that the background rate of PCT is relatively low in the populations in these latter studies and that finding an association would require study of larger populations of patients.
The cause of the probable association between hepatitis C and PCT has been debated extensively in the literature. Some contend that hepatitis C serves to trigger attacks of PCT in predisposed patients, but this theory remains to be conclusively proven. Alternatively, others have hypothesized that patients with PCT are somehow more susceptible to hepatitis C infection. In either case, the probable linkage between these two diseases suggests that patients with PCT should be tested for hepatitis C, and that their treatment may be augmented by considering concomitant therapy for hepatitis C.
Pancreatic fat necrosis
First identified in 1883 by Chiari, the term pancreatic fat necrosis describes the association of skin nodules with pancreatic disease. The dermatologic lesions can be painless or painful and tend to appear first on the legs followed by the buttocks and trunk. [62] Initial presentation may include pruritus of lower extremities with associated splotchy erythema. Progression to a nodular component is associated with tenderness. The nodules have been described to spontaneously drain white creamy exudate, after which the lesion heals with hyperpigmentation and local scar formation. The pathologic correlate includes the presence of subcutaneous fat necrosis together with ghostlike calcified fat cells containing thick shadowy walls and no nucleus.
The presence of these fluctuant lesions on the skin surface has been used to identify underlying pancreatic disease. [63, 64] Often, it is the skin eruption that leads the patient to seek medical care in the first place. Pancreatic fat necrosis occurs in up to 65% of patients with pancreatic cancer and in up to 22% of patients with pancreatitis. More than half the patients with pancreatic cancer–associated subcutaneous fat necrosis have the acinar form. [65] The presence of subcutaneous fat necrosis along with pancreatic disease is a poor prognostic indicator; one series of 27 patients demonstrated that all 8 patients with associated pancreatic carcinoma and 8 of the 19 patients with pancreatitis died as a result of their disease.
The development of subcutaneous nodules in cases of pancreatic disease may derive from the release of pancreatic lipolytic enzymes, a hypothesis confirmed by the biochemical discovery of persistently elevated serum lipase and trypsin levels 100-fold in one case report. The relief of pancreatic insult by endoscopic retrograde cholangiopancreatography (ERCP) in another recent case report not only halted serum amylase release but also resolved the symptoms of panniculitis. [66]
Lichen planus
Lichen planus is an idiopathic inflammatory disorder of the skin and mucous membranes known to be associated with a variety of liver diseases. The skin lesions are purple, polygonal, flat-topped papules that are usually pruritic. A characteristic finding on close (through a lens) examination is the presence of white or gray linear markings known as Wickham striae. They may be found anywhere on the skin, although certain sites, such as the wrists, ankles, shins, lower back, and genitalia, are most commonly affected.
The mucous membranes are also frequently affected [67] and can be the only site involved. The lesions are most frequently white reticulated papules that form a lacelike pattern. Commonly affected sites include the buccal mucosa and lateral surfaces of the tongue. Precipitating factors for oral lesions include mechanical trauma from dental procedures and dental prostheses, heat and irritation from tobacco products, and oral habits to include lip and cheek chewing. Both cutaneous and oral lesions exhibit Köbner phenomenon, occurring at sites of previous trauma. Nail dystrophy is also common, including the presence of pterygia and longitudinal ridges. Treatment for both the skin and mucous membrane lesions involves the use of local or systemic steroids, specially formulated oral preparations of topical immunomodulators, and systemic medications, many of which are immunosuppressives, for treatment-resistant cases.
The pathogenesis of lichen planus is not clear, but several authors have postulated an association with both primary biliary cirrhosis (PBC) and chronic hepatitis B and C. [68, 69, 70, 71, 72, 73, 74] The association of PBC is particularly compelling because both diseases involve immunopathologic mechanisms that are not yet fully understood. However, not all authors have agreed that these associations occur at any more than the expected random frequency.
Trousseau syndrome
This syndrome was first described by Armand Trousseau in 1865, when he observed migratory thrombophlebitis in cancer patients. Two years later, he diagnosed the syndrome on himself and eventually succumbed to gastric cancer. In the modern clinical setting, Trousseau syndrome has been used to describe a spectrum of disease manifestations: from the classic migratory thrombophlebitis, to arterial emboli, to verrucous endocarditis, to any kind of coagulopathy occurring in any kind of malignancy. [75] The classic definition of migratory thrombophlebitis and the dermatologic findings therein are described.
Migratory thrombophlebitis presents as inflamed, red-colored, and painful linear lesions, representing a vasculitis caused by clot formation. These clots form, resolve, and then form again elsewhere, migrating across the trunk and extremities of the patient. [75]
Of these lesions, 25-50% have a neoplastic association. This is most strongly noted with pancreatic cancer but can occur in other adenocarcinomas. The mechanism by which malignancies cause these lesions is multifactorial and involves the disruption of the mechanisms regulating the clotting cascade. [76]
Trousseau syndrome usually resolves with the treatment of the underlying malignancy. However, heparin-based anticoagulation may be used to prevent further clot formation. Vitamin K antagonists, such as warfarin, are not effective treatment agents.
Necrolytic migratory erythema
Necrolytic migratory erythema presents as annular (ring-shaped) patches of erythema that blister, erode, and crust over. When observing a patient, all stages of the lesions, from patches, to vesicles, to crusts, may be seen synchronously. The lesions can be pruritic or painful and are commonly distributed over the perioral region, lower abdomen, buttocks, groin, and lower legs. [77]
In the literature, up to 70% of necrolytic migratory erythema is associated with glucagonomas, which are glucagon-secreting tumors of the pancreatic alpha cells. However, necrolytic migratory erythema can also be associated with liver disease and intestinal malabsorption. [78] The pathogenesis of this condition is unknown.
Necrolytic migratory erythema usually resolves with treatment of the underlying disease. However, owing to the poor prognosis of glucagonomas, the outcome of this condition is also poor.
Necrolytic acral erythema
Necrolytic acral erythema (NAE) was first described in 1996 in Egypt by el Darouti and Abu el Ela. [79] NAE manifests as a psoriasiform eruption with well-circumscribed violaceous-to-erythematous plaques with thick adherent scale. The lesions are associated with burning or pruritus and are limited to an acral distribution. In most cases, NAE is associated with hepatitis C infection, [80] although several cases have occurred in patients without hepatitis C. [81, 82] Overall, it is a rare entity, with one study showing a prevalence 1.7% in patients with hepatitis C. [83] The pathophysiology of this condition is not fully known, but the reports of NAE without hepatitis C infection support an idea the condition may be a result of zinc dysregulation, rather than a result of hepatitis C infection itself.
Dermatology and the Small Intestine
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disorder affecting approximately 1 in 10,000 live births. PJS is characterized by the presence of mucocutaneous hyperpigmentation together with GI polyposis. Clinical diagnosis requires histologic identification of intestinal hamartomatous polyps in conjunction with 2 of 3 additional clinical criteria that include small bowel polyposis, mucocutaneous melanotic pigmentation, and a family history of PJS. The skin findings first appear in infancy or early childhood and involve blue-brown macular lesions of 1-10 mm. The lesions most commonly are found on the lips (95% of patients) [84] and buccal mucosa (83%). Other frequently affected sites include the palms of the hands, fingers, nose, gingiva, eyelids, and hard palate. Over time, the mucosal lesions tend to persist, while the cutaneous lesions fade away. Ruby and argon lasers have been used successfully to eradicate the mouth pigmentation.
GI manifestations of PJS involve the presence of multiple hamartomatous polyps occurring most commonly in the jejunum (see the image below). Other sites include the stomach, duodenum, ileum, and colon. Symptoms may include abdominal pain, bleeding, rectal prolapse, or obstruction due to intussusception. In addition, 2-3% of patients develop GI carcinoma during their lifetimes, a frequency that has led to the recommendation of upper and lower endoscopy every 2 years in patients with PJS, with removal of all large polyps. [85, 86]
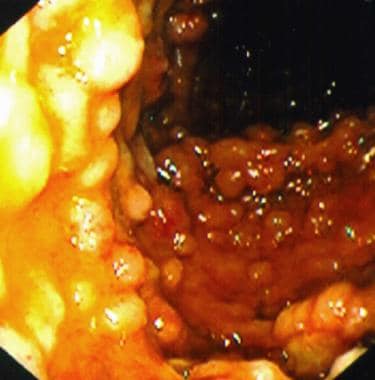 Multiple large intestinal polyps in a patient with Gardner syndrome. Courtesy of Christina Surawicz, MD, Harborview Medical Center, Seattle, Wash.
Multiple large intestinal polyps in a patient with Gardner syndrome. Courtesy of Christina Surawicz, MD, Harborview Medical Center, Seattle, Wash.
Care of these patients also involves regular surveillance for cancers involving the breast, ovary, testicle (Sertoli cell testicular tumors, leading to associated gynecomastia), cervix, thyroid, and other tissues. [87] These recommendations are supported by a recent long-term cohort study of 34 patients with PJS that demonstrated both a disproportionate number of noncutaneous cancers (26 cases, 16 non-GI tract, relative risk 9.9) and an unusually young age of diagnosis of these carcinomas (average age 39.4 y). Patients with a clinical diagnosis of PJS should have ongoing surveillance of all these organ systems.
Henoch-Schönlein purpura
This small vessel vasculitis manifests clinically as palpable purpura, arthralgias, abdominal symptoms, and glomerulonephritis. [88, 89] Males are affected twice as frequently as females, with an overall incidence of 14 cases per 100,000 population. The diagnosis usually is made in early childhood, but it also can present in infancy or adulthood. [90] The disease is believed to be due to the deposition of immunoglobulin A (IgA)–mediated immune complexes throughout the body. A variety of triggers for this process have been proposed, including insect bites, seasonal allergens, upper respiratory tract infections, various medications, and foods.
The skin abnormalities are present in all patients at some point in their disease process and involve the presence of palpable purpuric lesions found most commonly on the buttocks and legs. The lesions may appear in groups and may be vesicular or ulcerating in some cases. Definitive diagnosis can be obtained through direct immunofluorescence of early lesions (aged < 24 h), revealing a perivascular deposition of IgA. Renal findings in patients with this disease include glomerulonephritis with the presence of proteinuria, microscopic hematuria, and often red cell casts. Musculoskeletal pain can be diffuse and severe in some cases, although less than 20% of patients initially present with this symptom. Adults, in comparison to children, seem to more commonly present with symmetric arthralgias and arthritis of knees and ankles.
Gastroenterologic symptoms, which are found in more than 50% of patients with Henoch-Schönlein purpura (HSP), include colicky abdominal pain, diarrhea/constipation, occult or occasionally overt bleeding, and rarely intussusception or perforation. [91] These symptoms may appear simultaneously with the skin lesions, or they may appear several days to weeks earlier. Endoscopic evaluation may demonstrate marked generalized redness, raised lesions, multiple ulcerations, or diffuse erosive lesions. [92]
The use of corticosteroids has been reported to be helpful in reducing the GI symptoms, although glomerulonephritis and bowel infarction may be better treated with cyclosporine and mycophenolate mofetil. [93, 94] The natural course of the disease is complete resolution over weeks to months. However, the presentation of HSP differs significantly with age; older patients exhibit much more severe renal and extrarenal manifestations than younger patients.
Dermatitis herpetiformis
This uncommon blistering disorder is characterized by the development of small vesicular lesions found in a symmetric distribution of both upper and lower extensor surfaces and the scalp. First described in 1884 by Louis Duhring, MD, the usual age of onset of dermatitis herpetiformis (DH) is in the third decade of life. Most patients experience severe pruritus as a result of the skin lesions, although some are almost completely asymptomatic. Pruritus can lead to significant excoriation and secondary changes, leading to misdiagnosis of atopic dermatitis. The characteristic direct immunofluorescence finding is granular deposition of IgA within the dermal papillae on direct immunofluorescence. Patients may have elevated serum IgA antibodies against transglutaminase 2 and 3. [95]
Twenty percent of patients with DH have a clinically overt celiac disorder, while almost 100% of patients with DH have demonstrable pathologic changes of celiac disease on small intestinal biopsy. [96, 97] Both DH and celiac disease have been found to share class II HLA allele sites in immunogenetic studies, a finding confirmed clinically by case reports of monozygotic twins afflicted by both diseases.
The clinical manifestations of the celiac disease (also known as gluten-sensitive enteropathy and celiac sprue) are caused by the inability to absorb gluten from the diet. Patients experience weight loss, diarrhea, bloating, and steatorrhea. Chronic problems with malabsorption eventually may lead to iron or folate-deficient anemia states. Therapy involves the adoption of a gluten-free diet, with avoidance of foods such as wheat, barley, and rye. The initiation of therapy with dapsone or sulfapyridine results in a dramatic improvement in both GI and dermatologic symptoms; a prompt improvement following therapy can confirm the diagnosis.
Blue rubber bleb nevus syndrome
This rare disorder is characterized by the combination of cutaneous vascular malformations and GI bleeding due to the presence of vascular malformations. [98, 99] A clinical syndrome first described by Bean in 1958, transmission may be sporadic or autosomal dominant. The clinical manifestations most often present in birth or early childhood, although some cases may not be identified until adulthood. The skin lesions can number from 1 to more than 100 and may come in 3 forms:
-
Nontender soft nodules that when compressed leave behind a blue empty sac that refills rapidly with blood (blue rubber nipple)
-
Blue-black punctate tender macular lesions widely distributed on the extremities and trunk
-
Large hemangiomas (up to 10 cm in diameter) that may interfere with important limb or organ function
The GI manifestations of the blue rubber bleb nevus syndrome involve the presence of vascular malformations found most frequently in the small intestine and colon (although lesions have been identified in the mesentery, lung, liver, eye, and CNS.) The malformations project into the gut lumen and resemble the nodular skin lesions in appearance. Clinically, these malformations usually cause occult bleeding, although frank melena, hematochezia, or even intussusception may occur. The treatment is largely dependent upon the the disease course. For patients with mild blood losses over time, therapeutic intervention can involve monitoring, iron replacement, and blood transfusions when needed. In severe cases, treatment may require endoscopic therapy with bipolar electrocautery or YAG laser, or even surgical resection of affected areas.
Dermatology and the Large Intestine
Familial adenomatous polyposis (Gardner syndrome)
Gardner syndrome is an autosomal dominant disorder involving the triad of epidermal cysts, osteomas, and adenomatous GI polyposis. [100] Originally described by Gardner in 1953, the disease is now known to involve a single gene defect on chromosome 5. [101] The epidermal cysts are found in more than 50% of patients and tend to appear in decreasing likelihood of occurrence on the lower extremities, face, scalp, and upper extremities. The age of onset is the early teenage years, and they are almost always multiple. Cutaneous cysts frequently predate intestinal polyps. Osteomas, located most frequently in the mandible or maxilla, occur in at least 75% of patients with Gardner syndrome. Other associated findings include desmoid tumors (visceral and nonvisceral types), pilomatricomal cysts, dental abnormalities, and pigmented ocular fundal lesions termed congenital hypertrophy of the retinal epithelium.
The GI manifestation of Gardner syndrome involves the presence of adenomatous polyps, which are believed to occur in 100% of patients at some point in their lifetime. The polyps are first noted at an average age of 22 years and tend to occur in large numbers (>100) in any part of the colon. These lesions, which also can be found in the duodenum around the ampulla of Vater, eventually progress to carcinoma in almost all patients if left untreated. Therapy involves regular colonoscopy and excision of polyps.
Muir-Torre syndrome
Muir-Torre Syndrome (MTS) is an autosomal dominant disorder with variable expressivity and is characterized by skin lesions with colonic malignant potential. [102] Described separately by Muir and Torre in 1967, the syndrome associates sebaceous neoplasms with an increased propensity for colorectal carcinoma. The defect has been attributed to a DNA mismatch repair gene mutation, a discovery that now allows physicians to screen relatives of affected patients. There are diverse skin findings in MTS, including sebaceous adenoma, sebaceous epithelioma, sebaceous carcinoma, basal cell carcinoma with sebaceous cell differentiation, and keratoacanthomas. [103]
Unlike the universal presence of polyps in Gardner syndrome, patients with MTS have colonic polyposis in only approximately half the cases. However, visceral malignancies develop in a large proportion of patients, most frequently in the colon (51% of all primary tumors). Other sites of carcinoma include the larynx, duodenum, ileum, stomach, uterus, ovary, ureter, kidney, and bladder. The malignancies associated with MTS tend to be less aggressive than those unassociated with the syndrome, leading to a better prognosis than one might otherwise suppose given the malignancy. Treatment of patients with MTS involves regular screening for GI and genitourinary cancers.
Cowden disease (multiple hamartoma syndrome)
Cowden disease is a rare disease of autosomal dominant inheritance that is characterized by hamartomas of various tissues. Dermatologic findings include trichilemmomas (ie, skin colored papules around facial orifices), acral keratoses, and oral papillomas. The disease is associated with a variety of malignancies, including breast, thyroid, endometrial, cervical, and colon cancer.
Colonic polyps occur in up to 95% of patients with Cowden disease. [104] The most common sites of polyposis are the colon and rectum, although polyps have been documented in the esophagus, stomach, gallbladder, and small bowel.
A review of the 67 patients with Cowden disease who have undergone endoscopic evaluation over the past 30 years [105] demonstrates that most polyps discovered are nonadenomatous, although approximately 24% were adenomatous. Of particular concern are the 12 patients (18%) who had adenomatous colonic and rectal polyps, an established risk factor for colorectal cancer. Although the precise risk of colon cancer in patients with Cowden disease has not been firmly established, all patients should receive a thorough initial GI workup with follow-up care as appropriate.
Bannayan-Riley-Ruvalcaba syndrome
This is a rare autosomal dominant genodermatosis with the classic triad of macrocephaly, genital lentiginosis, and intestinal polyposis. Given overlap with Cowden syndrome with phenotypic expression and similar allelic syndromes (both with PTEN mutations), both diseases are collectively referred to as PTEN hamartoma-tumor syndrome. Mucocutaneous findings include vascular malformations, lipomatosis, speckled lentigines of the penis and vulva, facial acanthosis nigricans‒like lesions, and multiple acrochordons.
Similarly to Cowden disease, patients can have prominent diffuse hamartomatous polyposis and vascular lipomatous hamartomas manifesting as tender or painful, rapidly enlarging, aggressive subcutaneous and/or visceral lesions. Prominent hamartomatous polyposis occurs in 35-45% of cases of Bannayan-Riley-Ruvalcaba syndrome but is not associated with an increased risk of malignancy. Polyps can be located anywhere along the GI tract, with a preference for colonic or rectal localization. Presentation of polyposis may not manifest until middle age, requiring routine follow-up visits with a yearly hemoglobin test and fecal occult blood test. During childhood, symptoms include massive watery diarrhea, abdominal pain, painless rectal bleeding, and chronic anemia. The polyps can be quite large, leading to intussusception and intestinal obstruction.
Cronkhite-Canada syndrome
This tetrad of diffuse macular hyperpigmentation, alopecia, nail atrophy, and GI polyposis initially was described in 2 patients by Cronkhite and Canada in 1952. [106, 107] This extremely rarely identified syndrome has been further defined with the collection of more than 50 cases from the literature. The average age of onset is 59, with a range of ages of 31-86 years. The hyperpigmented macules and plaques (85% of patients) most often occur on the upper extremities but may be diffusely distributed. Nail dystrophy (affecting 90% of patients) is seen in all fingers and toes and is characterized by onycholysis and a unique pattern of normal nail in the shape of an inverted triangle that borders up against dystrophic nail. The alopecia (>95% of patients) described with this disease is of rapid onset, with progression from patchy hair loss to eventual complete loss of hair. [108]
GI symptoms include diarrhea, significant weight loss, and abdominal pain. The presence of hamartomatous polyps is quite common, with transformation to malignancy occurring in some cases.
Cronkhite-Canada syndrome is fatal in about one half of patients, usually as a result of malnutrition or persistent diarrhea. Therapy involves nutritional support and careful observation for metabolic derangements over the course of the illness. [109] One case report noted a temporal association with the initiation of ranitidine therapy and resolution of clinical symptoms in one patient, although the mechanism of response to therapy in this patient is unclear.
Inflammatory bowel disease
The two main diseases under the umbrella of inflammatory bowel disease (IBD) are Crohn disease (CD) and ulcerative colitis (UC). IBD is associated with a vast array of dermatologic findings, which are discussed below. Since there is large overlap between the skin findings in CD and UC, the diseases are considered together in this discussion.
CD is characterized clinically by symptoms of fever, abdominal pain, fatigability, and diarrhea that may or may not be bloody. The disease most often is diagnosed in patients aged 15-35 years, although it has been described in people of all ages. Histologically, the inflammatory lesions of CD extend through all layers of the gut lining and may extend into the mesentery or local lymphatics. The disease may appear in all areas of the GI tract from the mouth [110] to the anus and often is characterized by skip lesions, areas of the bowel that are completely free of inflammation. Although UC affects patients in similar age groups, this disease is more likely to result in bloody diarrhea and abdominal distension. The pathologic appearance of this disease demonstrates continuous colonic involvement from the rectum to the junction with the terminal ileum, with the inflammation confined to the mucosal layer. Diagnosis of both diseases generally is made easily with a combination of sigmoidoscopy and barium-assisted radiologic studies.
Cutaneous manifestations of IBD are quite common, with various studies suggesting skin involvement in 9-19% of patients with UC and 9-40% in patients with CD. Younger age and female sex have demonstrated statistically significant associations with cutaneous findings in IBD. [111] Cutaneous findings may precede IBD diagnosis or symptoms; thus, it is important to consider further workup when a patient presents with common IBD-associated dermatologic findings that recur or are refractory to treatment. [112] The cutaneous findings can be categorized into four main groups based on etiology. The groups have disease-specific cutaneous manifestations, reactive cutaneous manifestations, associated cutaneous disorders, and secondary cutaneous manifestations due to complications of IBD or IBD therapy (see Table 2 below).
Table 2. Dermatologic Manifestations of Inflammatory Bowel Disease (Open Table in a new window)
Disease-Specific Cutaneous Manifestations |
Metastatic CD |
Reactive Cutaneous Manifestations |
Erythema nodosum, pyoderma gangrenosum, Sweet syndrome, leukocytoclastic vasculitis, aphthous ulcers, pyostomatitis vegetans |
Associated Cutaneous Disorders |
Psoriasis, hidradenitis suppurativa, vitiligo, acquired epidermolysis bullosa |
Secondary Cutaneous Disorders Due to Complications of IBD or IBD Therapy |
Acrodermatitis enteropathica (see below), skin cancers, psoriasis |
The most common skin findings are described below.
Fissures
Although an infrequent phenomenon in patients with UC, fissures of the skin in patients with CD are common, and often quite uncomfortable. The most common area is the perineum, with particular involvement of the perianal area. The National Cooperative Crohn's Disease study demonstrated that patients with colonic involvement are significantly more likely to have perianal fissures than those with CD affecting other areas of bowel (40% compared vs 25%). As with other patients with perianal fissures, treatment with topical nitroglycerin or injected botulinum toxin can be quite effective.
Oral lesions
Mouth findings of patients with CD include findings such as angular cheilitis, aphthous ulceration, or mucosal cobblestoning. Metastatic CD lesions present as linear ulcerations that appear like a knife-cut both on the skin and the mucosa. One study found an incidence of oral findings in patients with CD of approximately 0.5%.
Pyoderma gangrenosum
Pyoderma gangrenosum (PG) is classically an ulcerative disease, but it can present in several forms, including ulcerative, bullous, pustular, and superficial. The most common form of PG is the ulcerative type, but notable is that when the pustular variant is seen, it is most commonly found in patients with IBD. PG is found in approximately 5% of patients with UC and about 1% of those with CD. Classic ulcerative PG presents as a painful, ulcerative lesion with a well-defined, rolled, dusky-appearing border (see the image below). The lesions start as small pustules, which subsequently expand to form the larger noninfectious ulcer. PG is frequently associated with pathergy (development of the condition at areas of trauma, surgery, or breakdown of the skin barrier). Patients who develop PG following surgery are at statistically increased risk of developing it again, following a second surgery. [113] PG can occur as a solitary entity, but up to 50% of patients with PG have an underlying systemic disorder such as IBD.
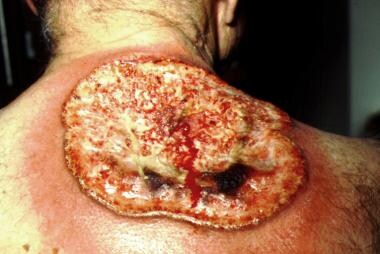 Pyoderma gangrenosum. Note the rolled-up, edematous, and undermined border with the surrounding halo of bright-red erythema. The base of the ulcer contains a fibrinopurulent exudate. This ulcer evolved from a "pimple" over a period of just a few weeks.
Pyoderma gangrenosum. Note the rolled-up, edematous, and undermined border with the surrounding halo of bright-red erythema. The base of the ulcer contains a fibrinopurulent exudate. This ulcer evolved from a "pimple" over a period of just a few weeks.
PG is most commonly associated with UC; therefore, an evaluation of the colon should be considered in all patients presenting with PG. Treatment of the IBD or other underlying causative illnesses may lead to an improvement in the skin lesions. Importantly, PG does not necessarily parallel the intestinal disease; therefore, patients may need further therapy to treat the PG. Some patients require systemic treatment with oral corticosteroids, cyclosporine, mycophenolate mofetil, antitumor necrosis factor (TNF) therapy, and other immunosuppressive agents. Studies demonstrated efficacy of anti-TNF therapy in treating refractory PG in IBD [114, 115] Chronic ulcers may respond to topical sodium cromoglycate.
Erythema nodosum
Erythema nodosum (EN) is the most common dermatologic manifestation of IBD, which presents as tender, nonulcerative, red nodules found most frequently on the lower legs. EN lesions usually parallel the intestinal disease, which provides helpful information on the overall disease. Histologically, the lesions are characterized by septal panniculitis. EN is associated with a variety of conditions, including medications, malignancy, and infections. In one large series of patients, 7% of patients with UC had EN, with a less frequent association in CD. Treatment targeted at the underlying IBD usually resolves skin lesions. EN does not typically precede the onset of IBD, and it rarely occurs in the quiescent phases of IBD. [116] In cases of EN not responsive to IBD treatments, additional therapy, such as prednisone, may be necessary.
Vulvar lesions
Vulvar lesions in CD can range from nonspecific ulcerations to exophytic lesions, with the presence of granulomatous inflammation and/or dilated lymphatic channels on biopsy. Most identified lesions include fistulae, abscesses, vulvar edema, hypertrophic lesions, and knifelike ulcerations at the inguinal folds, which are the most characteristic lesions and essentially pathognomonic for CD. [117] The majority of cases are due to fistulae arising from the digestive tract (anal, rectal, enteric fistulas); however, cutaneous CD of vulva is a recognized entity, although much rarer. Treatments include corticosteroids, metronidazole, azathioprine, and anti-TNF therapies. Surgical treatment is usually reserved for medically refractory cases. [118]
Medication-induced complications
Immunosuppressive medications used to treat IBD can lead to an increase in the risk of skin cancers, particularly in patients on thiopurines and anti-TNF therapy. [119] Regular skin cancer screening in this patient population may be warranted. Additionally, anti-TNF therapy can both treat and induce psoriasis. Studies have shown up to 3.5% of patients on anti-TNF therapy for IBD may develop psoriasis. Females appear to be at higher risk for developing psoriasis, and the most commonly affected areas are the palms, soles, and scalp. [120, 121]
Dermatology and Nutritional Deficiencies
Scurvy
Scurvy is a clinical syndrome of vitamin C deficiency. Vitamin C (ascorbic acid) is an essential dietary nutrient, whose deficiency can lead to impaired collagen synthesis and connective-tissue disorders. In the United States, vitamin C deficiency is seen in malnourished individuals from alcohol and drug abuse, impoverishment, and/or restricted diet, or in infants and elderly persons who are not getting adequate vitamin C in their diet. Symptoms typically appear within a few months of deficient intake. It is characterized by multiple cutaneous symptoms, as well as arthralgia, weakness, malaise, depression, and neuropathy.
Dermatologically, the cutaneous manifestations of scurvy are perifollicular lesions (hyperkeratotic papules, hemorrhage), corkscrew hairs, petechiae and ecchymosis, splinter hemorrhages of the nails, spongy bleeding of the gingiva, poor wound healing, and alopecia. [122] Notably, perifollicular lesions, petechiae, and ecchymosis occur more often below the hip, where there is greater hydrostatic pressure. [123]
Prognostically, scurvy is fatal if left untreated. However, it can be easily treated with foods containing vitamin C (eg, citrus fruits, spinach, kale, broccoli) or with over-the-counter supplements. Breast milk provides an adequate amount of vitamin C for infants and children. In infant formula, the pasteurization process can destroy vitamin C contained in milk, [124] and improper heating and storage can also destroy vitamin C that is added to the formula as part of the pasteurization process; thus it is important to follow recommended preparation and usage guidelines when using it.
Pellagra
The dietary deficiency of niacin (vitamin B-3) or tryptophan (which is converted into niacin) causes pellagra. [125] Symptomatically, pellagra is characterized by the 4 D’s: diarrhea, dermatitis, dementia, and death, usually presenting sequentially in that order. [126] It is seen in malnourished patients and in patients on certain medications (isoniazid, chloramphenicol, fluorouracil, and mercaptopurine).
Pellagra has several characteristic cutaneous manifestations, including photosensitive dermatitis, perineal lesions, and thick darkened skin over bony prominences. The photosensitive dermatitis appears as a red and glossy rash typically distributed over the dorsal hands and feet. The dermatitis may be painful or may cause a burning sensation, but it is rarely itchy. [123]
The treatment of pellagra involves oral niacin or nicotinamide. Owing to the association of pellagra with malnutrition, supplements of other nutrients, such as protein and other vitamin B complexes, should also be considered. [127]
Acrodermatitis enteropathica
Acrodermatitis enteropathica is a rare autosomal recessive disorder that impairs dietary zinc absorption in the jejunum and ileum. [128] Because zinc is better absorbed from breast milk than formula, the condition typically presents in infants several weeks after breastfeeding is discontinued, and earlier if infants are fed on formula. It is characterized by diarrhea, inflammatory rash, and hair loss.
The cutaneous signs of acrodermatitis enteropathica, as its name suggests, typically involve the extremities (acral areas), but also commonly involve the perioral and perianal areas. The lesions initially present as scaly, erythematous patches and plaques similar to atopic dermatitis, but progress to vesicles, crusts, erosions, and pustules. Additionally, other signs of disease may include nail plate inflammation (paronychia) and dystrophy, diffuse hair loss (telogen effluvium), mucous membrane involvement, and frequent skin infections. [129]
Patients with this condition are treated with lifetime zinc supplements. In general, clinical improvement occurs days to weeks after treatment initiation. The scaling and erosions present on the skin may also be treated symptomatically with warm compresses and moisturization.
-
Oral Kaposi sarcoma in a patient with AIDS. Note the characteristic purple hemorrhagic papules coalescing into an irregular plaque.
-
Multiple large intestinal polyps in a patient with Gardner syndrome. Courtesy of Christina Surawicz, MD, Harborview Medical Center, Seattle, Wash.
-
Pyoderma gangrenosum. Note the rolled-up, edematous, and undermined border with the surrounding halo of bright-red erythema. The base of the ulcer contains a fibrinopurulent exudate. This ulcer evolved from a "pimple" over a period of just a few weeks.
-
Koilonychia. Note the double concavity (longitudinal and transverse) of the nails.
-
Scleroderma affecting the hands. Note the taut appearance of the skin and the curved nails.
-
Acanthosis nigricans (AN) in a patient with pancreatic cancer. Note the papillomatous appearance of the axillary skin. This patient had previously been diagnosed with typical AN related to diabetes mellitus. After a long period of stability, the AN became much more severe and involved other parts of his skin, including the eyelids and scalp, prompting the search for malignancy.
-
Sister Mary Joseph nodule in a patient with gastric carcinoma. Note the shiny, reddish, telangiectatic group of papules in the umbilicus.
-
Arteriovenous malformations as seen on CT scan in a patient with Osler-Weber-Rendu syndrome. The patches of hyperdensity within the liver are the result of previous embolization procedures.
-
Telangiectases in the gastric mucosa of a patient with Osler-Weber-Rendu syndrome. The lesions can be seen most prominently at the 2-o'clock position proximally and the 3-o'clock position distally. Note the prominent red color of the lesions. Although these particular lesions appear flat, some GI telangiectasias may be slightly elevated. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
-
Postcricoid web in a patient with Plummer-Vinson syndrome. Note the 2 small openings within the web at the 2- and 6-o'clock positions, representing a significantly compromised proximal esophageal lumen. Reprinted with permission from Gastrointestinal Endoscopy, Second edition, Gower Medical Publishing, New York, 1991.
-
Patient with porphyria cutanea tarda.
-
Patient with dermatitis herpetiformis; often these patients are afflicted with celiac sprue.
-
The lacy, white reticulated lesions in this patient are consistent with lichen planus.
Tables
Dermatologic Manifestation |
GI Abnormality |
Disorder |
|
Periorificial granulomas [3] |
Malabsorption |
Crohn disease |
|
Koilonychia |
Esophageal webs |
Plummer-Vinson syndrome |
|
Liver disease |
Hemochromatosis |
||
Palmoplantar keratoderma |
Esophageal carcinoma |
Howel-Evans syndrome |
|
Acral rash |
Bazex syndrome [4] |
||
Telangiectasia [5] |
Esophagitis |
Scleroderma |
|
GI bleeding |
Hereditary hemorrhagic telangiectasia |
||
Cirrhosis |
Liver disease secondary to alcohol or other factors |
||
Vesicles/blisters/erosions |
Esophageal webs |
Epidermolysis bullosa |
|
Esophageal erosion |
Pemphigus vulgaris |
||
Pyloric atresia |
Junctional epidermolysis bullosa |
||
Hepatitis |
Porphyria cutanea tarda |
||
Malabsorption |
Dermatitis herpetiformis and celiac sprue |
||
Velvety hyperpigmented plaques, tripe palms, mucosal hyperplasia |
Gastric cancer |
Malignant acanthosis nigricans |
|
Yellowish papules, "chicken skin" |
GI bleeding |
Pseudoxanthoma elasticum |
|
Eruptive xanthomas |
Pancreatitis |
Primary biliary cirrhosis |
|
Biliary cirrhosis |
Sister Mary Joseph nodule |
||
Panniculitis |
Pancreatic |
Pancreatitis Pancreatic cancer |
Pancreatitis Pancreatic cancer |
Erythema nodosum |
Malabsorption |
Inflammatory bowel disease |
|
Skin tumors |
Multiple cysts |
GI polyps Colon cancer |
Gardner syndrome |
Sebaceous neoplasms, multiple keratoacanthomas |
Muir-Torre syndrome |
||
Trichilemmomas |
Cowden syndrome |
||
Hyperpigmentation |
Mucosal macular |
GI polyps Upper GI cancer |
Peutz-Jeghers syndrome |
Diffuse macular |
GI polyps GI cancer |
Cronkhite-Canada |
|
Diffuse |
Hepatomegaly Cirrhosis Hepatocellular carcinoma |
Hemochromatosis |
|
Plaques, acral |
Hepatitis |
Necrolytic acral erythema |
|
Lichenoid papules/plaques |
Hepatitis |
Lichen planus |
|
Hepatomegaly [6] Hepatic lesions |
Sarcoidosis |
||
Papules that heal with atrophic scars |
Perforation |
Malignant atrophic papulosis [7] |
|
Vascular malformation |
GI bleeding |
Blue rubber bleb nevus syndrome |
|
Ulcers with undermined borders |
Malabsorption |
Pyoderma gangrenosum and inflammatory bowel disease [8] |
|
Palpable purpura |
Abdominal pain |
Henoch-Schönlein purpura |
|
Plaques, intertriginous |
Malabsorption |
Acrodermatitis enteropathica |
|
Disease-Specific Cutaneous Manifestations |
Metastatic CD |
Reactive Cutaneous Manifestations |
Erythema nodosum, pyoderma gangrenosum, Sweet syndrome, leukocytoclastic vasculitis, aphthous ulcers, pyostomatitis vegetans |
Associated Cutaneous Disorders |
Psoriasis, hidradenitis suppurativa, vitiligo, acquired epidermolysis bullosa |
Secondary Cutaneous Disorders Due to Complications of IBD or IBD Therapy |
Acrodermatitis enteropathica (see below), skin cancers, psoriasis |







