Practice Essentials
Pityriasis lichenoides is a rare cutaneous disorder of unknown etiology. Pityriasis lichenoides encompasses a spectrum of clinical presentations ranging from acute papular lesions that rapidly evolve into pseudovesicles and central necrosis (pityriasis lichenoides et varioliformis acuta or PLEVA) to small, scaling, benign-appearing papules (pityriasis lichenoides chronica or PLC). [1, 2] Historically, the term Mucha-Habermann disease has generally referred only to PLEVA, however, some have included PLC as well. A rare febrile ulceronecrotic variant has been reported, which is a severe form of PLEVA with high fever and marked constitutional symptoms. Lesions may self-involute and resolve completely over weeks, or new lesions occasionally may appear in crops, waxing and waning spontaneously for months to years thereafter.
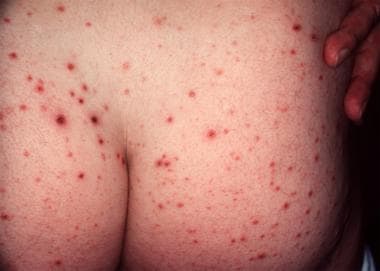 Typical hemorrhagic crusted papules of pityriasis lichenoides et varioliformis acuta.
Typical hemorrhagic crusted papules of pityriasis lichenoides et varioliformis acuta.
Prognosis
No clear consensus has been formed regarding duration of the disease, but most cases tend to resolve over time. Patients must be told that lesions may take time to resolve and that the duration of the disease cannot be predicted. The skin-limited form of pityriasis lichenoides is a self-limited disease.
The febrile-ulceronecrotic variant may arise de novo or from a preexisting case of pityriasis lichenoides. Rare reports of death from the febrile-ulceronecrotic variant have been attributed to secondary pulmonary thromboembolism, pneumonia, cardiac arrest, and sepsis, among others.3 Ulceronecrotic pityriasis lichenoides et varioliformis acuta (PLEVA) can lead to scarring.
Diagnostics
Laboratory workup largely is a function of the acuity of the disease. A patient presenting with febrile ulceronecrotic pityriasis lichenoides et varioliformis acuta (PLEVA) requires an entirely different approach than a patient presenting with pityriasis lichenoides chronica (PLC).
The following laboratory tests address both implicated causes of Mucha-Habermann disease and other disorders in the differential diagnosis; tailor the workup to each patient's presentation:
-
Antistreptolysin O titers
-
EBV IgM/IgG viral capsid antigen and nuclear antigen antibody
-
Erythrocyte sedimentation rate
-
Hepatitis B surface antigen, antisurface antibody, and anticore IgM
-
Hepatitis C virus antibody
-
HIV screening
-
Monospot or heterophil antibody test
-
Rapid plasma reagin
-
Throat cultures
-
Toxoplasma Sabin-Feldman dye test, enzyme-linked immunoassay, and indirect immunofluorescence/hemagglutination
Other tests
A punch biopsy of 4 mm or larger or shave biopsy should be strongly considered to ensure the diagnosis and rule out lymphomatoid papulosis.
Dermoscopy of an Indian girl with pityriasis lichenoides chronica of the trunk revealed structureless areas with yellowish-orange to light brown color with focal superficial scales and multiple scattered dark brown to black granules. A few scattered vessels were also observed. Focal areas of hypopigmentation were noted on the periphery of the lesions. [3]
T-cell gene rearrangement studies to test for clonality may aid in the diagnosis of a lymphoma. It should be noted, however, that benign dermatoses also may show T-cell gene restriction and that the discovery of a clone is not a sine qua non of a lymphoma diagnosis. Several studies have shown a significant portion of pityriasis lichenoides cases have a T-cell clone. [4, 5]
Histologic findings
Ackerman has established histopathologic criteria for fully developed lesions of PLEVA and PLC. [6] Early lesions in both variants are smooth, since areas of parakeratosis initially are overlain by a normal cornified layer with a basket-woven appearance.
PLEVA lesions are characterized by a wedge-shaped superficial and deep dermal lymphohistiocytic infiltrate with intravascular margination of neutrophils, a confluent parakeratotic scale crust, thinning of the granular layer, basilar necrosis of keratinocytes, vacuolar interface dermatitis with a lymphocyte in nearly every vacuole, erythrocyte extravasation, and dermal edema.
The presence of plasmacytoid dendritic cells might be helpful in differentiating PLEVA from lymphomatoid papulosis. [7]
Rare cases of γδ T-cell–predominant disease may mimic more aggressive lymphomas histologically. [8, 9]
PLC lesions are characterized by a superficial dermal infiltrate, focal parakeratosis, preservation of the granular layer, and focal disappearance of the dermal-epidermal interface.
Treatment
Also see Long-Term Monitoring and Medication.
Large ulcerations found in the febrile ulceronecrotic variant of pityriasis lichenoides et varioliformis acuta (PLEVA) require local wound care. [10] Infected lesions may be treated with topical mupirocin and sterile dressing changes twice daily.
No randomized controlled trials of the use of medications have been performed in Mucha-Habermann disease. Since the disease tends towards self-resolution, evaluation of treatments without adequate controls cannot result in useful recommendations. A number of open trials have reported success with light therapy and oral medications, particularly tetracycline antibiotics. [11] Phototherapy is generally the most effective approach, with methotrexate reserved for severe or refractory disease. [12, 13, 14, 15, 16] Hrin et al reported their experience with methotrexate in both PELVA and PLC and found it to be of benefit for both conditions. [17]
Surgical care
Two cases in the literature have reported a tonsillectomy in patients with chronic tonsillitis or high ASO titer and PLC and PLEVA, respectively. [18, 19] One patient's skin disease resolved within days of the tonsillectomy, while the other's persisted for over 5 years.
Pathophysiology
Mucha-Habermann disease is not a vasculitic process despite reports of immunoglobulin and complement deposition in vessels. Fibrin is not present in the walls of vessels, and thrombi are not found in the lumen. A cell-mediated mechanism has been proposed based on a T-lymphocytic infiltrate with a cytotoxic/suppressor phenotype, diminished epidermal Langerhans cells, and a reduction of the CD4/CD8 ratio. CD30 (Ki-1) cells, which are associated with large cell lymphoma, have been identified in the infiltrate of both lymphomatoid papulosis and Mucha-Habermann disease, leading some authors to view this as a self-limited self-healing lymphoproliferative disease. [20, 21] One study suggests that pityriasis lichenoides is a form of a T-cell dyscrasia, based on the presence of intraepithelial atypical lymphocytes, phenotypic abnormalities, and TCR-gamma rearrangements. [4]
Etiology
The exact etiology of PLC and PLEVA remains elusive; many cases resolve without the discovery of an identifiable culprit. Consideration has been given to the possibility of a low-grade or self-limited lymphoproliferative disorder, a response to an infectious agent, or an inflammatory reaction to an unknown epitope.
A number of acute exanthems (eg, Mucha-Habermann disease, pityriasis rosea, acute lichen planus, guttate psoriasis, erythema multiforme) are believed to be caused by a hypersensitivity reaction to infectious agents. Familial outbreaks, clustering of cases, and comorbid symptoms have been used to support these relationships in Mucha-Habermann disease, although clear causality is lacking. Elevations of pathogen-specific antibody titers also have been offered as proof of causality, but such immunologic responses may represent epiphenomena caused by nonspecific immune responses to unknown pathogens. The most commonly reported associated pathogens are Epstein-Barr virus (EBV), Toxoplasma gondii, and human immunodeficiency virus (HIV).
Two studies implicate EBV as an etiologic factor in Mucha-Habermann disease. The cases indicate that EBV may be a trigger in PLEVA, but neither study necessarily illustrates well-characterized comorbid EBV-mediated disease. Note the following:
-
In 1977, Boss et al reported a cluster of 10 cases seen over 1 year, in which eruptions were clinically consistent with PLEVA. [22] Of these, 4 demonstrated elevated immunoglobulin G (IgG) complement-fixing antibodies to EBV. During resolution of the eruption, 3 of 4 patients demonstrated 4-fold or greater decrements in antibody titers.
-
In 1989, Edwards et al described a child with a 3-week history of migratory arthralgias, monoarticular arthritis, acute pharyngitis, otitis media, and fevers to 104ºF. [23] The girl developed a vesicular eruption localized primarily to the extremities, which clinically and histopathologically was consistent with Mucha-Habermann disease, with the exception of necrotic fibrin thrombi in the superficial and mid dermis. A Monospot test result was positive, and acute and convalescent serologies were consistent with a reactivation of EBV. Liver function tests were within normal limits. The patient's condition improved with treatment using oral tetracycline.
Elevated Toxoplasma gondii titers have been demonstrated in some patients with Mucha-Habermann disease. Despite an absence of clinical infection in case reports and series, more than 80% of primary Toxoplasma infections are asymptomatic, and toxoplasmosis cannot necessarily be dismissed as a causative agent. Note the following:
-
In 1969, Andreev et al were the first to suggest a link between toxoplasmosis and a recurrent PLEVA-like skin eruption in a patient with positive Toxoplasma serologies. [24] Cutaneous lesions reportedly responded favorably to pyrimethamine.
-
In 1972, Zlatkov and Andreev reported 11 patients with PLC and found that test results were positive for toxoplasmosis in 6 patients (55%) using complement-fixations test, intradermal test with toxoplasmin, and Sabin-Feldman dye test. [25] Patients in whom test results were seropositive responded favorably to pyrimethamine, while no improvement was noted in the cutaneous lesions of 3 patients in whom results were seronegative.
-
In 1987, Rongioletti et al described a patient who presented with acute onset of cutaneous lesions and histopathologic findings consistent with PLEVA. [26] Serologic examination demonstrated enzyme-linked immunosorbent assay positivity for IgG and immunoglobulin M (IgM) and weak positive indirect fluorescence test results for IgM (1:16). Giemsa stain on the biopsy specimen failed to demonstrate Toxoplasma cysts. Spiramycin treatment was initiated, and lesions subsided over a few weeks. Convalescent serologies failed to demonstrate IgM 2 months later, although the authors still concluded that Toxoplasma species may have caused the cutaneous eruption.
-
In 1997, Nassef and Hammam reported 22 patients diagnosed clinically and histopathologically with PLC and 20 healthy control subjects. [27] Clinical examination for signs of toxoplasmosis only revealed axillary lymphadenopathy in 2 patients. Eight patients with PLC (36%) had a positive serodiagnosis by indirect hemagglutination versus 10% in the control group, and this difference was statistically significant. Using indirect immunofluorescence antibody tests, the difference was 36% versus 15%, respectively, but the difference was not statistically significant. All 22 patients with PLC were treated with pyrimethamine and trisulfapyrimidine, and lesions in 5 of 8 patients with seropositive results cleared completely within 2 months. None of the patients with seronegative results responded to treatment.
The first association between Mucha-Habermann disease and HIV infection was reported in 1991 by Ostlere et al. [28] A patient with asymptomatic disease and a CD4+ T-cell count of 208 cells per microliter, diagnosed 6 months previously, presented with lesions consistent clinically and histopathologically with PLEVA. Note the following:
-
In 1997, Smith et al reported a series of 5 patients with HIV infection in the early stage of disease, with CD4+ T-cell counts exceeding 200 cells per microliter and/or absolute lymphocyte counts within normal limits. [29] The authors suggested that PLEVA serves as a marker of early–to–mid stage HIV disease.
-
In 1998, Griffiths reported a patient who presented with a severely pruritic, erythematous, papular eruption that worsened as the CD4+ T-cell count fell from 200 to 20 cells/μ L. [30] Biopsy confirmed PLC, and the disease progressed to febrile ulceronecrotic PLEVA. Dramatic improvement was attained using cyclosporine, and mild PLC-like lesions remained on maintenance doses. On saquinavir and lamivudine, the viral load became undetectable with a concomitant rise in the CD4+ count and a complete resolution of skin lesions. That the inherent immunologic dysregulation of HIV may play a role in Mucha-Habermann disease has been suggested.
In addition to EBV, Toxoplasma gondii, and HIV, a number of other infectious agents have been implicated. The following observations are provocative but may be chance associations. Note the following:
-
Case reports have suggested that parvovirus B19 and adenovirus can trigger Mucha-Habermann disease.
-
Herpes simplex has been associated with onset of the disease.
-
One case report also describes resolution of PLC after tonsillectomy, with throat cultures yielding Staphylococcus aureus and group A beta hemolytic streptococci.
-
Piamphongsant similarly found coagulase-positive staphylococci on throat cultures in 4 of 10 patients, with some improvement of cutaneous lesions using oral tetracycline. [31]
-
Freeze-dried live attenuated measles vaccine administered by injection has been associated with Mucha-Habermann disease [32] .
-
A 12-year-old boy presented with PLEVA five days following influenza vaccination. [33]
Interestingly, with the increased usage of antitumor necrosis factor-α agents, several reports of PLC induction in patients have been published. [34, 35, 36, 37] These events remain rare and in patients with underlying inflammatory conditions such as Crohn disease. Additionally, it is not clear if a preceding illness was found in these immunosuppressed patients.
Epidemiology
Frequency
The incidence of Mucha-Habermann disease in the United States has not been reported. In approximately 44,000 patients seen over 10 years in 3 catchment areas in Great Britain, 17 cases of PLEVA were diagnosed.
Race-,sex-,and age-related information
All races are affected. A racial predisposition has not been reported.
A male predominance has been reported in the pediatric population and in patients presenting with febrile ulceronecrotic Mucha-Habermann disease.
Most patients present during the first 3 decades of life. Studies of children have shown a variable age of onset from 3-15 years, with a mean age of 9.3 years. The chronic form is more common in children. [38]
Prognosis
A case series of 22 children revealed a mean duration in PLEVA of 1.6 months to complete resolution and a mean duration in PLC of 7.5 months. The natural tendency of the disease is to remit spontaneously, but some cases may wax and wane over years. Disease duration may be longer in adults. A rare severe variant of PLEVA presents with a sudden eruption of diffuse coalescent necrotic ulcerations associated with high fever. [39] Patients may develop complications such as interstitial pneumonitis, abdominal pain, malabsorption, central nervous system involvement, bacteremia, sepsis, and rheumatic manifestations. T-cell receptor clonal rearrangements of lymphocytic infiltrates have been detected in patients with PLEVA. Occasional cases (< 2%) have been reported to evolve into cutaneous lymphoma, although some reports may have represented misdiagnosis of lymphomatoid papulosis. [40]
-
Typical hemorrhagic crusted papules of pityriasis lichenoides et varioliformis acuta.
-
Close-up view of typical lesions of pityriasis lichenoides et varioliformis acuta.
-
Scaling papules of pityriasis lichenoides chronica.
-
Close-up view of typical pityriasis lichenoides chronica lesions. Note papules in different stages of evolution and the scale with frosted-glass appearance in the lower right-hand corner.