Background
Aplasia cutis congenita (ACC) [1, 2, 3] is a heterogenous group of disorders characterized by the absence of a portion of skin in a localized or widespread area at birth. First reported by Cordon in 1767, aplasia cutis congenita manifests as a solitary defect on the scalp in 70% of cases, but it may sometimes occur as multiple lesions. Although most commonly seen on the scalp, aplasia cutis congenita can affect any part of the body, including the trunk and limbs.
See 13 Common-to-Rare Infant Skin Conditions, a Critical Images slideshow, to help identify rashes, birthmarks, and other skin conditions encountered in infants.
The lesions of aplasia cutis congenita are noninflammatory and well-demarcated, ranging widely in size. They may be circular, oval, linear, or stellate in configuration. Aplasia cutis congenita occurring in a blaschkoid distribution has also been reported in 3 cases. [4, 5, 6] At birth, the lesions may have already healed with scarring or may remain superficially eroded to deeply ulcerated, occasionally involving the dura or the meninges. Defects in the skin that form early in gestation may heal before delivery and appear as an atrophic, membranous, [7] bullous, [8] or parchmentlike scar with associated alopecia, whereas less mature defects present as ulcerations. The membranous type of aplasia cutis congenita is most common.
Most lesions of aplasia cutis congenita occur on the scalp vertex just lateral to the midline, but defects may also occur on the face, trunk, or limbs, sometimes symmetrically. The defect may involve only the epidermis and upper dermis, resulting in minimal alopecic scarring, or it may extend into the deep dermis, subcutaneous tissue, or rarely periosteum, skull, and dura. Extension to deeper structures should be suspected in large, irregular lesions of aplasia cutis.
Aplasia cutis congenita is most often a benign isolated defect, but it can be associated with other physical anomalies or malformation syndromes.
Frieden [1] created a classification system for aplasia cutis congenita consisting of 9 groups based on the number and location of the lesions and the presence or absence of associated malformations:
Group 1
This is scalp aplasia cutis congenita without multiple anomalies. [9] Nearly 86% of all solitary lesions occur on the scalp. A collar of hair is often seen around the defect, particularly with membranous aplasia cutis. It can be autosomal dominant [10] or sporadic. [11]
Group 2
This is scalp aplasia cutis congenita with limb anomalies. [12] Adams-Oliver syndrome [13, 14, 15, 16, 17] is a distinct disorder in which distal limb reduction abnormalities are found in association with solitary midline scalp defects. Adams-Oliver syndrome exhibits both autosomal dominant and autosomal recessive patterns of inheritance. In recent years, mutations in EOGT and DOCK6 have been identified as causes for autosomal recessive Adams-Oliver syndrome, and mutations in DLL4,ARHGAP31, RBPJ, and NOTCH1 have been observed in autosomal dominant forms of the syndrome. [18, 19, 20, 21, 22, 23] In this group of aplasia cutis congenita, the scalp lesions tend to be large. The most common limb malformation is hypoplastic or absent distal phalanges, but the severity of limb anomalies ranges from minor defects such as an absent nail or broad fingertip to more severe involvement. Limb anomalies are usually asymmetric and more commonly involve the lower extremities. [24] Other anomalies may include cutis marmorata telangiectatica congenita, hemangiomas, cranial arteriovenous malformation, congenital heart defects, skin tags, supernumerary nipples, and woolly hair.
Group 3
This is scalp aplasia cutis congenita with epidermal and sebaceous (organoid) nevi, [25, 26] also involving the scalp usually adjacent to the aplasia cutis. Some patients have also had ophthalmic and neurologic findings typical of epidermal nevus syndrome, including seizures, intellectual disability, corneal opacities, and eyelid colobomas. SCALP syndrome has been proposed as an entity to describe the constellation of nevus Sebaceus, Central nervous system malformations, Aplasia cutis congenita, Limbal dermoid, and Pigmented nevus. [25] Inheritance is sporadic.
Group 4
This is aplasia cutis congenita overlying deeper embryologic malformations. [27, 28, 29, 30] Examples include meningomyelocele, porencephaly, leptomeningeal angiomatosis, cranial stenosis, spinal dysraphism, gastroschisis, and omphalocele. A hair collar is often present in the scalp lesions overlying neural tube defects. Intracranial arteriovenous malformations and fistulas have also been reported in association with scalp aplasia cutis congenita in rare cases. [31] The inheritance pattern in this group varies with the associated underlying condition. There is often a need for abdominal wall defect repair with this type of aplasia cutis congenita. [32]
Group 5
This is aplasia cutis congenita associated with fetus papyraceous or placental infarct. [33, 34, 35, 36, 37, 38, 39] Fetus papyraceous is found at the time of delivery and results from the death of a twin fetus in the late first or early second trimester. The surviving fetus is affected with extensive truncal and limb aplasia cutis congenita in a linear or stellate configuration but is usually otherwise normal.
Group 6
This is aplasia cutis congenita associated with epidermolysis bullosa (EB), [40, 41, 42] , also referred to as Bart syndrome. Aplasia cutis congenita can be seen with any type of EB (simplex, junctional, or dystrophic). Many reports describe aplasia cutis congenita usually occurring on the lower extremities. A subgroup includes the association of pyloric or duodenal atresia, ureteral stenosis, renal abnormalities, craniofacial abnormalities, nail dystrophy, and aplasia cutis congenita.
Group 7
This is aplasia cutis congenita localized to the extremities without EB. [43, 44, 45] At least two families have been reported in which multiple members have had extensive aplasia cutis congenita on the pretibial lower extremities and the dorsal aspects of the hands and the feet.
Group 8
This is aplasia cutis congenita due to teratogens. A few cases of aplasia cutis congenita have been linked to intrauterine infection with herpes simplex virus or varicella zoster virus or to exposure to methimazole [46, 47, 48, 49] in the treatment of maternal thyrotoxicosis during pregnancy. Imperforate anus has been associated with methimazole or carbimazole exposure during gestation.
Group 9
This is aplasia cutis congenita associated with malformation syndromes. [50, 51] Aplasia cutis congenita has been reported as a characteristic in many syndromes, [52, 53, 54, 55] including trisomy 13 (Patau syndrome) with large membranous scalp defects; 4p- (Wolf-Hirschhorn) syndrome with midline scalp defects; Setleis syndrome with bitemporal aplasia cutis congenita and abnormal eyelashes; Johanson-Blizzard syndrome with stellate scalp defects; focal dermal hypoplasia (Goltz syndrome); amniotic band disruption complex; oculocerebrocutaneous (Delleman) syndrome; scalp-ear-nipple syndrome (Finlay-Mark syndrome) [56] ; Kabuki syndrome [57] ; and 46XY gonadal dysgenesis. Reticulolinear aplasia cutis congenita on the face and neck is a distinctive cutaneous manifestation in several syndromes linked to band Xp22.
Pathophysiology
The exact pathophysiology of aplasia cutis congenita (ACC) is unclear. The most commonly accepted theory focuses on the tension that prevents the skin from converging during development in utero. Proposed mechanisms include intrauterine trauma, vascular compromise, infection, and medications. It has been theorized that stellate or angulated lesions in particular result from vascular abnormalities or intrauterine ischemia.
Aplasia cutis congenita is typically sporadic; however, autosomal dominant and, less commonly autosomal recessive, cases have also been reported. Mutations in the ribosomal GTPase BMS1 have been identified as one cause of autosomal dominant aplasia cutis congenita. [58] Familial aplasia cutis congenita on the scalp is generally nonmembranous, whereas membranous aplasia cutis congenita of the scalp is usually sporadic. [59]
Etiology
No unifying theory can account for all lesions of aplasia cutis congenita (ACC). Because this condition is the phenotypic result of more than one disease process, it is likely that more than one mechanism is involved. Mechanisms include genetic factors, teratogens (eg, methimazole, carbimazole, misoprostol, valproic acid), compromised vasculature to the skin, infections, neural tube defects, and trauma. Of particular note is the association of fetus papyraceous with bilaterally symmetric aplasia cutis congenita.
The proximity of scalp aplasia cutis congenita to the scalp hair whorl, which is thought to be the point of maximum tensile force during rapid brain growth, has led to the hypothesis that tension-induced disruption of the overlying skin occurs at 10-15 weeks of gestation when hair direction, patterning, and rapid brain growth occur. This may also explain the increased incidence of aplasia cutis congenita on the vertex scalp.
Early rupture of the amniotic membranes, forming amniotic bands, has appeared to be the cause of aplasia cutis congenita in several cases.
The bullous or membranous variants of aplasia cutis congenita reveal a distinct histologic pattern identical to those in encephaloceles and meningoceles. This supports a hypothesis that these types of aplasia cutis may represent the forme fruste of a neural tube closure defect.
Epidemiology
Frequency
Aplasia cutis congenita (ACC) is an uncommon anomaly of newborns. More than 500 cases have been reported since it was first described, but because of significant underreporting of this generally benign disorder, the precise frequency is unknown. For example, one estimate of incidence is approximately 3 cases in 10,000 births and another shows 0.3%. [60]
Race
No racial predilection is reported for aplasia cutis congenita.
Sex
Unless associated with an X-linked malformation syndrome, no sexual predilection exists in aplasia cutis congenita.
Age
Aplasia cutis congenita lesions are present at birth.
Prognosis
The prognosis for aplasia cutis congenita (ACC) is usually excellent. If the defect is small, recovery is uneventful, with gradual epithelialization and formation of a hairless, atrophic scar over several weeks. The average length of recovery for patients treated conservatively is 27.9 days. [61] Small underlying bony defects usually close spontaneously during the first year of life. Surgical repair of large or multiple scalp defects with excision and primary closure, if feasible, or with the use of tissue expanders and rotation of a flap, may be considered. Truncal and limb defects, despite their large size, usually epithelialize and form atrophic scars, which can later be revised if necessary.
If aplasia cutis congenita is associated with other anomalies, the prognosis is dependent on the severity of the associated abnormalities. Underlying or associated defects may significantly affect mortality and morbidity. Full-thickness defects of the scalp, skull, and dura are associated with a mortality rate of greater than 50%. Even large defects on areas other than the scalp usually heal well with conservative skin care using topical antibiotic ointment. The rare larger scalp defects are prone to complications of hemorrhage and infection, placing patients at risk for death. Extensive aplasia cutis congenita of the scalp may be associated with an increased risk of sagittal sinus thrombosis. For these reasons, surgical intervention may be required for large, full-thickness scalp defects.
Patient Education
Genetic counseling is advised for parents of affected individuals if there is a strong family history of aplasia cutis congenita (ACC) or if the affected children have aplasia cutis congenita in association with a malformation syndrome.
-
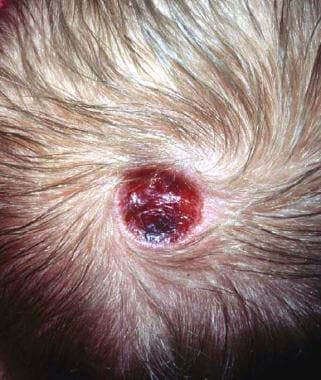
Aplasia cutis congenita on the scalp (most common location) shortly after birth.
-
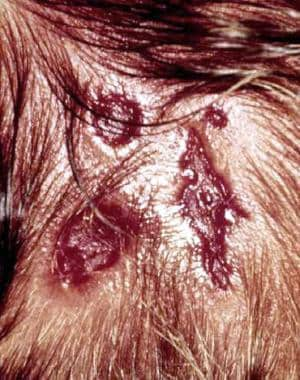
Triplet areas of aplasia cutis congenita are common in infants with trisomy 13.
-
This area of healed aplasia cutis congenita is located in an area of nevus flammeus. Note the collarette of coarser hair at the margin of the defect.
-
Extensive aplasia cutis congenita on the scalp, extending down to the skull.
-
Bilateral involvement of the lower extremities in aplasia cutis congenita associated with fetus papyraceous.





