Background
Bullous systemic lupus erythematosus is an autoantibody-mediated subepidermal blistering disease that occurs in patients with systemic lupus erythematosus (see the image below). [1, 2, 3, 4]
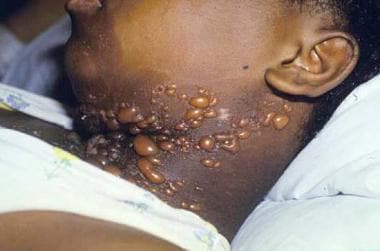 Tense vesiculobullous lesions on the neck of a patient with bullous systemic lupus erythematosus.
Tense vesiculobullous lesions on the neck of a patient with bullous systemic lupus erythematosus.
The diagnosis of bullous systemic lupus erythematosus requires the following elements (see Presentation and Workup):
-
Fulfillment of the American College of Rheumatology criteria for systemic lupus erythematosus (see also Systemic Lupus Erythematosus)
-
An acquired vesiculobullous eruption (see the image above)
-
Histologic evidence of a subepidermal blister and a predominantly neutrophilic dermal infiltrate
-
Direct immunofluorescence (DIF) microscopy demonstrating immunoglobulin G (IgG, with or without immunoglobulin A [IgA] and immunoglobulin M [IgM]) deposits at the basement membrane zone (BMZ)
-
Evidence of antibodies to type VII collagen via DIF or indirect immunofluorescence (IIF) on salt-split skin, immunoblotting, immunoprecipitation, enzyme-linked immunosorbent assay (ELISA), or immunoelectron microscopy [5]
All 5 criteria listed above are needed for a diagnosis of type 1 bullous systemic lupus erythematosus, whereas only the first 4 criteria are needed to diagnose type 2 (undetermined location of antigen or dermal antigen other than type VII collagen) and type 3 (epidermal antigen) bullous systemic lupus erythematosus. Type VII collagen, a component of anchoring fibrils, is also targeted in epidermolysis bullosa acquisita (EBA). However, unlike epidermolysis bullosa acquisita, bullous systemic lupus erythematosus tends to respond dramatically to treatment with dapsone. (See Differentials and Treatment.)
Not all blistering eruptions that occur in patients with lupus erythematosus represent bullous systemic lupus erythematosus as defined above. Vesiculobullous skin lesions can also develop as a result of extensive damage to the epidermal basal layer (and even suprabasal keratinocytes), due to an intense interface dermatitis in the setting of lupus erythematosus–specific skin disease. Such patients may present with a severe form of acute or subacute cutaneous lupus erythematosus (SCLE) that resembles erythema multiforme (Rowell syndrome) [6] or toxic epidermal necrolysis (TEN). (See Differentials.) [7, 8, 9]
Because epidermolysis bullosa acquisita and bullous systemic lupus erythematosus share the same target antigen, distinguishing between the 2 may be difficult. (See Etiology below.)
For patient education information, see Lupus (Systemic Lupus Erythematosus).
Etiology
The production of autoantibodies represents a central feature of systemic lupus erythematosus. For example, antinuclear antibodies (ANAs) are detected in almost all affected individuals. In addition to this general tendency, injury to the dermoepidermal junction by the interface dermatitis of cutaneous lupus erythematosus may expose new epitopes and precipitate the development of antibodies that specifically target the BMZ.
The autoantibody repertoire of systemic lupus erythematosus can include nonpathogenic and pathogenic anti-BMZ antibodies. Patients with nonbullous systemic lupus erythematosus often have circulating antibodies to various components of the BMZ (including bullous pemphigoid antigens 1 and 2), which may have a role in formation of the lesional lupus band (ie, granular antibody deposition at the BMZ in normal-appearing skin). [10]
Although the lupus band appears to colocalize with type VII collagen, the noncollagenous-1 (NC1) domain of this protein does not represent the target antigen for circulating antibodies in systemic lupus erythematosus patients without clinical blistering.
In patients with bullous systemic lupus erythematosus, antibodies directed at the BMZ likely mediate the blistering phenotype by directly interfering with adhesive connections at the dermoepidermal junction and through induction of complement-dependent inflammation that leads to tissue injury and dermoepidermal separation. Proteolytic damage caused by recruited neutrophils contributes to the latter process.
In patients with systemic lupus erythematosus without vesiculobullous eruptions, an increase of circulating antitype VII collagen antibodies may precede the onset of type 1 bullous systemic lupus erythematosus. The autoantibodies then increase after disease onset and decrease with remission of eruptions. [11] In type 1 bullous systemic lupus erythematosus (which accounts for most cases), antibodies against type VII collagen may weaken or block anchoring fibril-mediated connections between the lamina densa of the basement membrane and the papillary dermis.
In epidermolysis bullosa acquisita and bullous systemic lupus erythematosus, antigenic epitopes reside within the NC1 and NC2 domains of type VII collagen, which are localized to the lamina densa and the underlying dermis, respectively. [12] The cartilage matrix protein (CMP) subdomain of the NC1 domain has been shown to bind to antibodies from patients with epidermolysis bullosa acquisita and systemic lupus erythematosus, suggesting this subdomain serves as an immunodominant antigenic epitope. [13]
Subepidermal blisters can be induced in mice by the passive transfer of such autoantibodies from patients with epidermolysis bullosa acquisita, but not the Fab fragments alone. [14, 15] This demonstrates that human epidermolysis bullosa acquisita autoantibodies are pathogenic and that complement-mediated inflammation (which requires the Fc fragment) has an important role in the blistering process.
Antibodies recognizing bullous pemphigoid antigen 1, laminin-5, and laminin-6 have also been described in patients with bullous systemic lupus erythematosus. [16] While autoreactive helper T cells and dysregulation of regulatory T cells may play a role in other autoimmune blistering disorders, such as pemphigus and bullous pemphigoid, autoimmunity to type VII collagen has not been shown to involve aberrant regulatory T cell function. [17, 18, 19]
The term acute syndrome of apoptotic pan-epidermolysis (ASAP) has been proposed for the toxic epidermal necrolysis–like cutaneous injury pattern that can occur in settings of lupus erythematosus, acute graft versus host disease, pseudoporphyria, and the classic drug-hypersensitivity syndrome. [20] Fas-Fas ligand interactions have been implicated in the massive keratinocyte apoptosis that characterizes ASAP.
Toxic epidermal necrolysis–like cutaneous lupus erythematosus (which can be triggered by intensive ultraviolet exposure) must be differentiated from drug-induced toxic epidermal necrolysis occurring in a patient with lupus erythematosus. [21]
Genetic predisposition
Certain individuals may have a genetic predisposition to develop autoimmunity to BMZ antigens and to systemic lupus erythematosus. For example, epidermolysis bullosa acquisita, bullous systemic lupus erythematosus, and systemic lupus erythematosus are all associated with an increased prevalence of the HLA class II DR2 haplotype.
The antigen-presenting protein encoded by the DR2-associated DRB1*1501 allele (found in epidermolysis bullosa acquisita and bullous systemic lupus erythematosus patients) has been postulated to be involved in presenting type VII collagen epitopes to T lymphocytes.
Epidemiology
Bullous systemic lupus erythematosus accounts for 2-3% of cases of autoimmune subepidermal blistering disease, with an estimated incidence of fewer than 0.5 cases per million population per year.
Persons of any race can develop bullous systemic lupus erythematosus, but it occurs most frequently in African Americans. Bullous systemic lupus erythematosus affects women more often than men, reflecting the female preponderance in systemic lupus erythematosus.
Bullous systemic lupus erythematosus most often manifests in the second through fourth decades of life, but it has been reported in children and older adults. [22, 23, 24]
Prognosis
The course of bullous systemic lupus erythematosus is often remitting. Fortunately, unlike epidermolysis bullosa acquisita, treatment with dapsone is successful in most cases of bullous systemic lupus erythematosus. The disorder frequently resolves spontaneously in less than 1 year. In some cases, postinflammatory hypopigmentation may remain. [25]
The development of bullous systemic lupus erythematosus in patients with systemic lupus erythematosus does not typically lead to increased mortality. Morbidity depends on the extent of the eruption and the response to therapy.
Toxic epidermal necrolysis–like lupus erythematosus can result in considerable morbidity and even mortality if extensive areas of skin are denuded, especially in the context of severe systemic manifestations of lupus erythematosus.
-
Tense vesiculobullous lesions on the neck of a patient with bullous systemic lupus erythematosus.






