Background
Harlequin ichthyosis is the most severe form of autosomal recessive congenital ichthyosis. [1] Harlequin ichthyosis is characterized by a profound thickening of the keratin layer in fetal skin. The affected neonate is born with a massive, horny shell of dense, platelike scale and contraction abnormalities of the eyes, ears, mouth, and appendages, as is shown in the images below.
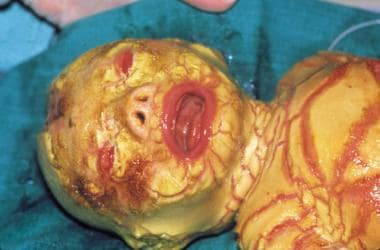
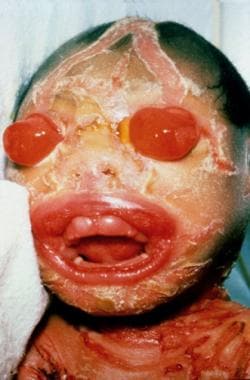
The term harlequin derives from the facial appearance and the triangular and diamond-shaped pattern of hyperkeratosis. The newborn's mouth is pulled wide open, mimicking a clown's smile.
Marked eclabium and ectropion are present secondary to the taut, unyielding skin. The ears may be absent or poorly developed. The arms, feet, and digits have flexion contractures and may be hypoplastic. The skin barrier is severely compromised, leading to excessive water loss, electrolyte abnormalities, temperature dysregulation, and an increased risk of life-threatening infection. The tight, armorlike scale can restrict respiration. Poor feeding and impaired intestinal absorption are common.
This disease primarily affects the skin. Other systems may be significantly compromised by the hyperkeratosis and concomitant deformities. Neonates are often born prematurely.
The underlying genetic abnormality in harlequin ichthyosis is a mutation in the lipid-transporter gene ABCA12 on chromosome 2.
Immunohistocytochemical examination of the skin reveals characteristic abnormalities in the structure of lamellar granules and in the expression of epidermal keratin.
In the past, harlequin ichthyosis was uniformly fatal. Improved survival has been achieved with intense supportive care and systemic retinoid therapy in the neonatal period. Patients who survive manifest a debilitating, persistent ichthyosis similar to severe congenital ichthyosiform erythroderma. [1, 2]
Other Medscape articles on ichthyosis include Hereditary and AcquiredIchthyosis Vulgaris, Lamellar Ichthyosis, X-Linked Ichthyosis, and Ichthyosis (ophthalmology focus).
Etiology
Genetic factors
Mutations in ABCA12, a gene that encodes adenosine triphosphate (ATP)-binding cassette transporter (ABC), subfamily A, member 12, in chromosome region 2q35, underlie this disorder. [3, 4] Patients with harlequin ichthyosis usually have functional null mutations in the ABCA12 gene. The mutations may be homozygous or compound heterozygous. [5] Missense mutations in ABCA12 result in milder autosomal recessive congenital ichthyosis phenotypes such as lamellar ichthyosis and congenital ichthyosiform erythroderma. [1, 4, 6]
The ABC superfamily of genes encodes proteins that transport a number of substrates across cell membranes. [6] ABCA12 encodes a transmembrane protein that mediates lipid transport.
This ABCA12 -mediated lipid-transfer system is essential to the transfer of lipids from the cytosol of the corneocyte into lamellar granules. Lamellar granules are intracellular granules that originate from the Golgi apparatus of keratinocytes in the stratum corneum. These granules are responsible for secreting lipids that maintain the skin barrier at the interface between the granular cell layer and the cornified layer in the upper epidermis. The extruded lipids are arranged into lamellae in the intercellular space with the help of concomitantly released hydrolytic enzymes. The lamellae form the skin's hydrophobic sphingolipid seal.
In harlequin ichthyosis, the ABCA12 -mediated transfer of lipid to lamellar granules is defective. The lamellar granules themselves are morphologically abnormal or absent. Normal extrusion of lipid from these granules into the extracellular space cannot occur, and lipid lamellae are not formed. This defective lipid "mortar" between corneocyte "bricks" results in aberrant skin permeability and lack of normal corneocyte desquamation. [7, 8, 9]
In vitro studies have demonstrated normalization of lipid transport when the wild-type ABCA12 gene is transferred to keratinocytes of patients with harlequin ichthyosis. [10]
One patient with a de novo deletion of chromosome arm 18q has been reported. [11]
Histopathologic, ultrastructural, and biochemical factors
Histopathologic, ultrastructural, and biochemical studies have identified several characteristic abnormalities in the skin of patients with harlequin ichthyosis.
The two main abnormalities involve lamellar granules and the structural proteins of the cell cytoskeleton. The relationship between mutations in the gene ABCA12 and abnormal lamellar granules is well documented. The pathophysiology of the other abnormalities documented below has yet to be elucidated.
Abnormal lamellar granule structure and function
The pivotal role of lamellar granules in maintaining a normal skin barrier is described in the Genetic factors bullet points at the beginning of this section.
All patients with harlequin ichthyosis have absent or defective lamellar granules and no extracellular lipid lamellae. Cultured epidermal keratinocytes from patients that carry ABCA12 mutations have demonstrated disturbed intracytoplasmic glucosylceramide transport. [6, 10] The lipid abnormality is believed to allow excessive transepidermal water loss. Lack of released lamellar granule enzymes prevents desquamation, resulting in a severe retention hyperkeratosis.
Abnormal conversion of profilaggrin to filaggrin
Profilaggrin is a phosphorylated polyprotein residing in keratohyalin granules in keratinocytes in the granular cell layer. During the evolution to the corneal layer, profilaggrin converts to filaggrin by means of dephosphorylation. Filaggrin allows dense packing of keratin filaments. Its subsequent breakdown into amino acids occurs prior to desquamation of the stratum corneum.
Some patients have a persistence of profilaggrin and an absence of filaggrin in the stratum corneum. A defect in protein phosphatase activity and subsequent lack of conversion of profilaggrin to filaggrin is hypothesized. [6]
Abnormal expression of keratin
Disturbed keratinocyte differentiation during fetal development is believed to play an important role in the pathogenesis of the harlequin ichthyosis phenotype at birth. [6] In utero studies have shown that expression of markers of late keratinocyte differentiation is highly dysregulated, suggesting a role for ABCA12 in keratinocyte differentiation. [12] Expression of the proteases kallikrein 5 and cathepsin D, which are required for normal desquamation, was found to be dramatically reduced. [12]
Keratin filament density is low in most patients. Expression of certain keratins is abnormal in some patients and normal in others. How this altered expression of structural proteins influences desquamation is uncertain.
Abnormal keratohyalin granules
Keratohyalin granules are identified by antifilaggrin antibodies and can be abnormal in some patients with harlequin ichthyosis. They can be large and stellate, small and rounded, or absent.
Epidemiology
Frequency
Approximately 200 cases of harlequin ichthyosis have been reported. [5] The incidence is calculated to be around 1 case in 300,000 births. [5]
Race
No racial predilection is known for harlequin ichthyosis. A higher incidence may be encountered in cultures where parental consanguinity is common. [13]
Sex
Sex distribution appears to be equal.
Prognosis
The mortality for harlequin ichthyosis rate is high, with worldwide figures approaching 50%. A review of 45 cases by Rajpopat et al found 25 survivors (56%), ranging in age from 10 months to 25 years. Twenty deaths (44%) occurred from day 1 to day 52 and were as likely to be caused by respiratory failure as fulminant sepsis. [13] A Japanese survey of 16 patients reported survival of 81.3% (13 of 16 patients). [14] Respiratory failure, fulminant sepsis, or a combination of both are the most common causes of death in affected newborns. [13]
Patient Education
Advise parents and caregivers that the baby’s appearance will improve after the neonatal period. Emphasize the need for attention to skin lubrication and for compliance with systemic therapy. Teach them to recognize signs of infection.
Congenital ichthyoses can have devastating medical and social consequences. Parents may wish to communicate with other families who have been similarly affected. Patient organizations (eg, The Foundation for Ichthyosis and Related Skin Types [FIRST]) are available in several countries to provide support to families.
-
Harlequin ichthyosis. Courtesy of Dr Bernice Krafchik.
-
Harlequin ichthyosis. Courtesy of Jason K Rivers, MD, FRCPC, and Dr Lawler.





