Background
Erythropoietic porphyria (EP) is a rare inborn error of porphyrin-heme synthesis inherited that is as an autosomal recessive trait. The inheritance of 2 mutant alleles for the gene encoding the enzyme uroporphyrinogen III synthase leads to accumulation of porphyrins of the isomer I type that are biologically useless but cause cutaneous photosensitivity characterized by blisters, erosions, and scarring of light-exposed skin.
Clinical manifestations can range from mild to severe. Chronic damage of skin, cartilage, and bones can cause mutilation. Hypertrichosis, erythrodontia, and reddish-colored urine are often present. Hemolytic anemia can be mild or severe, with resultant splenomegaly and osseous fragility.
The following is a selection of other porphyria-related articles:
Also see the figure below.
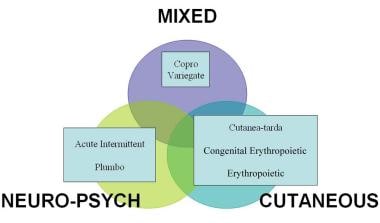 Clinical classification of porphyrias.
Clinical classification of porphyrias.
Pathophysiology
Erythropoietic porphyria is primarily a disorder of bone marrow heme synthesis. Deficient activity of the enzyme uroporphyrinogen III synthase in erythrocyte precursor cells causes a shift of the pathway away from the isomer III porphyrinogen production that can effect the end-product heme; isomer I porphyrinogens that cannot be used to form heme may be overproduced. The accumulated isomer I porphyrinogens are spontaneously oxidized to their corresponding porphyrins, which are water-soluble photosensitizers with a reddish hue.
These porphyrins are released from the maturing erythrocytes into the plasma and are excreted by renal mechanisms; urine with a port-wine color is produced. The interaction of excess porphyrins in the skin and light radiation causes photoxidative damage of biomolecular targets that is manifested as mechanical fragility and blistering that may result in severe scarring.
The hemolytic anemia of erythropoietic porphyria can cause hypersplenism in more serious cases. Hypertrophy of the bone marrow in such cases can lead to osseous fragility and pathologic fractures. Acral osteolysis and onycholysis may occur; bones and teeth are stained red by the deposition of porphyrin pigment. Ocular damage can lead to blindness. The photoactive nature of porphyrin molecules results in the bright pink fluorescence of these pigments in urine, teeth, and bones under Wood light illumination.
Etiology
Erythropoietic porphyria is caused by autosomal recessive inheritance of genes that encode abnormal uroporphyrinogen III synthase (UROS) enzyme protein. The resultant deficient activity of this enzyme leads to hemolytic anemia, cutaneous photosensitivity, and their complications. The mutation that causes the most severe deficiency of the enzyme uroporphyrinogen III synthase is C73R. The C73R mutation is responsible for more than 20% of the cases. [1] C73R mutations lead to premature degradation of the UROS enzyme by disturbing protein stability. [2]
The GATA gene family, a group of transcription factors, has a crucial role in normal human hematopoiesis. A mutation in GATA1, an X-linked transcription factor, has been reported in association with erythropoietic porphyria. [3]
Uroporphyrinogen I accumulates in the skin. With an excess of uroporphyrinogen and in the presence of 404-nm wavelengths of light, free radicals are formed leading to photodermatitis. Uroporphyrinogen fluoresces in ultraviolet light, causing the reddish discoloration seen in the teeth and sclera examined under a Wood light. Owing to the excretion of uroporphyrinogen in urine, it also fluoresces in ultraviolet light.
Epidemiology
Frequency
United States
A porphyria registry has only recently been established in the United States (American Porphyria Foundation); therefore, accurate figures are not yet available. Erythropoietic porphyria is rare; only several hundred cases have been reported worldwide.
International
Erythropoietic porphyria is reported in diverse populations. Only several hundred cases have been reported worldwide.
Race
No racial predilection is reported for erythropoietic porphyria.
Sex
Erythropoietic porphyria occurs in both males and females with approximately equal frequencies.
Age
Erythropoietic porphyria typically occurs in infants or young children; however, several adult-onset cases are reported.
Prognosis
Remarkable clinical variability exists in erythropoietic porphyria. Despite the limited treatments that are currently available, the prognosis is not invariably poor. With strict adherence to sun avoidance, scarring and mutilation can be minimized.
Normal life spans are possible in many cases. Most patients with erythropoietic porphyria survive into adulthood, with a life expectancy of 40-60 years.
-
Clinical classification of porphyrias.



