Background
The ectodermal dysplasias (EDs) comprise a large, heterogeneous group of inherited disorders that are defined by primary defects in the development of 2 or more tissues derived from embryonic ectoderm. The tissues primarily involved are the skin and its appendages (hair follicles, eccrine glands, sebaceous glands, and, nails) and teeth. Although Thurnam published the first report of a patient with ectodermal dysplasia in 1848, the term ectodermal dysplasia was not coined until 1929 by Weech. [1]
The ectodermal dysplasias are congenital, diffuse, and nonprogressive. To date, more than 192 distinct disorders have been described. The most common ectodermal dysplasias are X-linked recessive hypohidrotic ectodermal dysplasia (Christ-Siemens-Touraine syndrome), as shown in the image below, and hidrotic ectodermal dysplasia (Clouston syndrome).
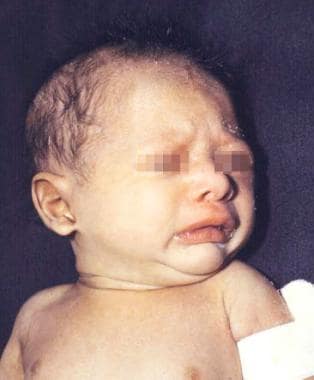 A newborn boy with anhidrotic/hypohidrotic ectodermal dysplasia syndrome showing generalized fine scaling and a history of intermittent fever.
A newborn boy with anhidrotic/hypohidrotic ectodermal dysplasia syndrome showing generalized fine scaling and a history of intermittent fever.
Current classification of ectodermal dysplasias is based on clinical features. Pure ectodermal dysplasias are manifested by defects in ectodermal structures alone, while ectodermal dysplasia syndromes are defined by the combination of ectodermal defects in association with other anomalies.
Freire-Maia and Pinheiro proposed the first classification system of the ectodermal dysplasias in 1982, [2] with additional updates in 1994 and 2001. [3, 4] Their original classification system stratified the ectodermal dysplasias into different subgroups according to the presence or absence of (1) hair anomalies or trichodysplasias, (2) dental abnormalities, (3) nail abnormalities or onychodysplasias, and (4) eccrine gland dysfunction or dyshidrosis.
Overall, the ectodermal dysplasias were classified into either group A disorders, which were manifested by defects in at least 2 of the 4 classic ectodermal structures as defined above, with or without other defects, and group B disorders, which were manifested by a defect in one classic ectodermal structure (1-4 from above) in combination with (5) a defect in one other ectodermal structure (ie, ears, lips, dermatoglyphics). Eleven group A subgroups were defined, each with a distinct combination of 2 or more ectodermal defects (eg, 2-4, 1-2-3, 1-2-3-4 from above). The group B disorders were indicated as 1-5, 2-5, 3-5, or 4-5 (from above). Visinoni tabulated a summary of the 186 defined ectodermal dysplasia syndromes classified as group A in 2009. [5] This classification was revised in 2014 to include 163 defined ectodermal dysplasia syndromes. [6]
With the recent identification of the causative genetic defect for a number of the ectodermal dysplasias, newer classification systems have been devised. In 2003, Lamartine reclassified the ectodermal dysplasias into the following 4 functional groups based on the underlying pathophysiologic defect: (1) cell-to-cell communication and signaling, (2) adhesion, (3) development, and (4) other. [7] Similarly, in 2001, Priolo and Laganà reclassified the ectodermal dysplasias into 2 main functional groups: (1) defects in developmental regulation/epithelial-mesenchymal interaction and (2) defects in cytoskeleton maintenance and cell stability. [8] Other classification systems categorize the ectodermal dysplasias based on defects in cell-cell communication and signaling, adhesion, transcription regulation, or development. [9]
Several ectodermal dysplasia syndromes may manifest in association with midfacial defects, mainly cleft lip, cleft palate, or both. The 3 most commonly recognized entities are (1) ectodermal dysplasia, ectrodactyly, and clefting (EEC) syndrome [10] ; (2) Hay-Wells syndrome or ankyloblepharon, ectodermal dysplasia, and cleft lip/palate (AEC) syndrome; and (3) Rapp-Hodgkin syndrome, all of which are caused by mutations in the TP63 gene. See the images below.
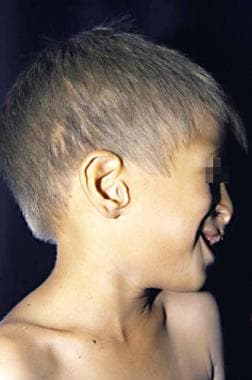 Ectodermal dysplasia, ectrodactyly, and clefting syndrome. Light-colored hair and scalp and earlobe defects are observed. Cleft lip and palate results in a characteristic nasal contour.
Ectodermal dysplasia, ectrodactyly, and clefting syndrome. Light-colored hair and scalp and earlobe defects are observed. Cleft lip and palate results in a characteristic nasal contour.
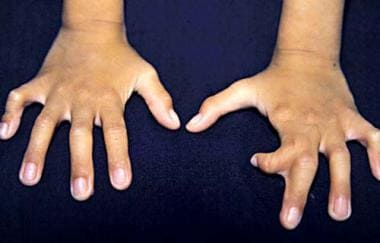 Ectrodactyly observed in an individual with ectodermal dysplasia, ectrodactyly, and clefting syndrome.
Ectrodactyly observed in an individual with ectodermal dysplasia, ectrodactyly, and clefting syndrome.
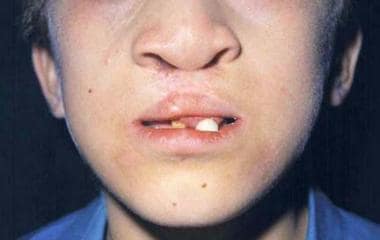 Typical cleft lip/palate and maxillary hyperplasia in a patient with Rapp-Hodgkin syndrome.
Typical cleft lip/palate and maxillary hyperplasia in a patient with Rapp-Hodgkin syndrome.
Pathophysiology
Ectodermal dysplasia results from the abnormal morphogenesis of cutaneous and/or oral embryonal ectoderm (ie, hair, nails, teeth, eccrine glands). In some forms, mesodermal abnormalities are also present. Characteristic features include the following:
-
Hair defects: A reduction in the number of hair follicles in conjunction with structural hair shaft abnormalities may be seen. Structural hair shaft abnormalities may result from aberrations in hair bulb formation and include longitudinal grooving, hair shaft torsion, and cuticle ruffling. Hair bulbs may be distorted, bifid, or small. [11]
-
Other secretory gland defects: Hypoplasia of the salivary, sebaceous, and lacrimal glands may occur. In some patients, mucous glands may be absent in the upper respiratory tract and in the bronchi, esophagus, and duodenum.
-
Dental defects: Abnormal morphogenesis or absence of teeth as well as enamel defects may occur. [13]
-
Nail dystrophy: Abnormal nail plate formation may result in brittle, thin, ridged, or grossly deformed nails.
Although some ectodermal dysplasia syndromes have no known genetic etiology, the number of ectodermal dysplasia syndromes with an identifiable genetic basis is increasing. In 2009, 64 genes and 3 chromosomal loci were associated with 62 ectodermal dysplasias. [5]
Key transcription factors and intracellular signaling pathways that have been implicated in the ectodermal dysplasias include the tumor necrosis factor (TNF)-like/TNV receptor signaling pathway, which involves ectodysplasin (EDA), the EDR receptor (EDAR), the EDAR-associated death domain (EDARADD); the WNT signaling pathway; the NF-kB signally pathway, which involves the NF-kB essential modulator (NEMO); and the transcription factor p63. [14]
Etiology
Ectodermal dysplasia results from the abnormal development of embryonic ectodermal structures. The genetic defects responsible for approximately 30 of the ectodermal dysplasias have been identified. However, a detailed understanding of the pathophysiology underlying most forms of ectodermal dysplasia with regards to the mechanisms by which the underlying genetic defects impact the growth and development of ectodermal structures is lacking.
X-linked recessive hypohidrotic ectodermal dysplasia (XL-HED or Christ-Siemens-Touraine syndrome) is caused by mutations in EDA, which encodes the ectodysplasin protein, a soluble ligand that activates the NF-kappaB and JNK/c-fos/c-jun signaling pathways. [15, 16] Ectodysplasin is important in promoting cell survival, growth, and differentiation. Using specialized techniques, including confocal imaging, phototrichogram analysis, and pilocarpineiontophoresis, a complete absence of eccrine ducts, a reduction in hair follicle units and hair follicle density, and a decreased growth rate of terminal hairs has been demonstrated in patients with XL-HED. [17]
Autosomal dominant and autosomal recessive hypohidrotic ectodermal dysplasia are caused by mutations in the DL gene, which encodes the EDA (ectodysplasin) receptor. [18] Autosomal recessive hypohidrotic ectodermal dysplasia may also result from mutations in the EDARADD gene, which encodes a protein that interacts with the EDA receptor. A heterozygous mutation in the TRAF6 gene has been described in a patient with hypohidrotic ectodermal dysplasia. [19]
Hidrotic ectodermal dysplasia (Clouston syndrome), which is an autosomal dominant disorder, is caused by mutations in GJB6, which encodes connexin 30, a component of intercellular gap junctions. [20]
EDA-ID and OL-EDA-ID are both caused by mutations in the NEMO gene, which encodes the regulatory subunit of the inhibitor-kappa kinase complex that regulates NF-kappaB activity. [21, 22, 23]
AEC (Hay-Wells) syndrome, Rapp-Hodgkin syndrome, EEC syndrome, limb-mammary syndrome, split hand-split foot malformation syndrome, and acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome are all caused by mutations in the TP63 gene. [24, 25] p63 is a transcription factor that regulates the activity of the tumor suppressor gene TP53.
The genetic defects underlying other ectodermal dysplasias are also known. Selected examples are as follows:
-
Keratitis, ichthyosis, deafness (KID) syndrome is caused by mutations in the GJB2 gene, which encodes connexin 26. [26]
-
Margarita Island ectodermal dysplasia is caused by mutations in the PVRL1 gene, which encodes nectin-1. [27]
-
Ectodermal dysplasia with skin fragility is caused by mutations in the PKP1 gene, which encodes plakophilin 1. [28]
-
Goltz syndrome (focal dermal hypoplasia) is caused by mutations in the PORCN gene. [29]
-
Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis are caused by mutations in the KRT14 gene, which encodes keratin 14. [30]
-
Pachyonychia congenita type I is caused by mutations in either KRT6A (keratin 6a) or KRT16 (keratin 16), while pachyonychia congenita type II is caused by mutations in either KRT6B (keratin 6b) or KRT17 (keratin 17). [31, 32, 33]
-
Incontinentia pigmenti is caused by mutations in NEMO, or NF-kB essential modulator. [40]
-
Anhidrotic ectodermal dysplasia with common variable immunodeficiency is caused by mutations in ORAI1, oral calcium release-activate calcium modulator 1. [41]
Epidemiology
Frequency
United States
The frequency of the different ectodermal dysplasias in a given population is highly variable. The prevalence of hypohidrotic ectodermal dysplasia, the most common variant, is estimated to be 1 case per 100,000 births.
International
Collectively, the prevalence of ectodermal dysplasia is estimated at 7 cases per 10,000 births.
Race
The ectodermal dysplasias have been reported most often in whites, but they have also been observed in persons of other races. Hidrotic ectodermal dysplasia has been reported in an extensive kindred of French-Canadian origin.
Sex
X-linked recessive hypohidrotic ectodermal dysplasia has full expression only in males. Female carriers outnumber affected men, but females show little or no signs of the condition. X-linked recessive anhidrotic ectodermal dysplasia (EDA) with immunodeficiency (EDA-ID) and the X-linked recessive syndrome of osteopetrosis, lymphedema, EDA, and immunodeficiency (OL-EDA-ID) are also seen exclusively in males. The remaining ectodermal dysplasias have no sexual predilection.
Age
Clinical recognition of ectodermal dysplasia varies from birth to childhood depending on the severity of symptoms and the recognition of associated complications. Many patients are not diagnosed until infancy or childhood, when dental anomalies, nail abnormalities, or alopecia become apparent.
AEC or Hay-Wells syndrome may manifest at birth as ankyloblepharon in association chronic scalp erosions. Hypohidrotic ectodermal dysplasia may manifest as scaling and erythema at birth. EEC syndrome and other related ectrodactyly syndromes (eg, acro-dermato-ungual-lacrimal-tooth [ADULT] syndrome and limb-mammary syndrome) are usually recognized at birth as a result of the characteristic limb deformities. Patients with anhidrosis or hypohidrosis may present in early infancy with recurrent episodes of hyperpyrexia.
Prognosis
The prognosis for most patients with ectodermal dysplasia is very good. Morbidity and mortality is related to the absence or dysfunction of eccrine and mucous glands. Beyond early childhood, life expectancy ranges from normal to slightly reduced.
If hypohidrosis is recognized in the neonatal period and managed appropriately, no evidence indicates that the life span for a person diagnosed with one of the common types of ectodermal dysplasia is shorter than average. Intermittent hyperpyrexia may occur in infants with decreased sweating. The mortality rate approaches 30%. Recurrent high fever may also lead to seizures and neurological sequelae.
Pharyngitis, rhinitis, cheilitis, and dysphagia may result from reduced numbers of functional mucous glands in the respiratory and gastrointestinal tracts.
Growth failure is common. [46]
Severe inflammatory scalp dermatitis with erosions may result in frequent infections and cause scarring alopecia in patients with AEC (Hay-Wells) syndrome and Rapp-Hodgkin syndrome.
Life span can be affected in some rare types of ectodermal dysplasia. For example, patients with ectodermal dysplasia with immunodeficiency are at risk for significant morbidity and mortality related to recurrent infections and failure to thrive.
Patient Education
Provide early guidance about temperature regulation, acceptable activities, and the risk of hyperpyrexia from febrile illnesses. Inform patients and families that antipyretics are not effective in treating hyperpyrexia associated with hypohidrosis. Instruct caregivers on proper skin care and monitoring for signs of infection in patients with chronic scalp dermatitis and erosions.
Additional information and support for families is available through the National Foundation for Ectodermal Dysplasias.
-
A newborn boy with anhidrotic/hypohidrotic ectodermal dysplasia syndrome showing generalized fine scaling and a history of intermittent fever.
-
Wrinkled, hyperpigmented skin around the eyes and everted lips are typical characteristics of anhidrotic/hypohidrotic ectodermal dysplasia syndrome.
-
Typical cleft lip/palate and maxillary hyperplasia in a patient with Rapp-Hodgkin syndrome.
-
Abnormal hair shaft showing pili torti and a longitudinal groove (pili canaliculi) from a patient with Rapp-Hodgkin syndrome.
-
Hands of father and son with Rapp-Hodgkin syndrome. Nails have the same characteristics; they are brittle, thin, and dystrophic.
-
Ectodermal dysplasia, ectrodactyly, and clefting syndrome. Light-colored hair and scalp and earlobe defects are observed. Cleft lip and palate results in a characteristic nasal contour.
-
Ectrodactyly observed in an individual with ectodermal dysplasia, ectrodactyly, and clefting syndrome.









