Practice Essentials
Mucopolysaccharidoses (MPSs) are a group of lysosomal storage diseases, each of which is produced by an inherited deficiency of an enzyme involved in the degradation of acid mucopolysaccharides, now called glycosaminoglycans (GAGs). These diseases are autosomal recessive, except for mucopolysaccharidosis type II, which is X-linked. There are currently 13 identified MPSs, 2 of which were discovered in the 2020s. [1]
In addition to the Medscape orthopedics article Mucopolysaccharidosis, the following are pediatrics articles on mucopolysaccharidoses:
-
Mucopolysaccharidosis Type II (Hunter syndrome)
-
Mucopolysaccharidosis Type III (Sanfilippo syndrome)
-
Mucopolysaccharidosis Type IV (Morquio syndrome, see images below)
-
Mucopolysaccharidosis Type VI (Maroteaux-Lamy syndrome)
-
Mucopolysaccharidosis Type VII (Sly syndrome)
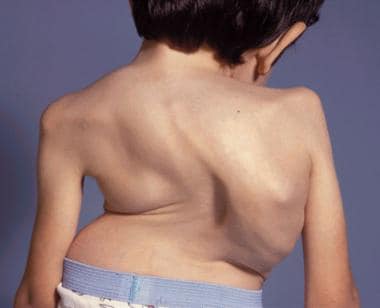 An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
 A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
Patient education
The following Web sites are resources for patients and the medical community: The National MPS Society, National Organization for Rare Disorders, and National Tay-Sachs & Allied Diseases Association.
Diagnosis
See Workup.
Treatment
See Treatment.
Pathophysiology
GAGs are long, linear polysaccharide molecules composed of repeating dimers, each of which contains a hexuronic acid (or galactose in the case of keratan sulfate) and an amino sugar. The large proteoglycan molecules made up of protein cores, and GAG branches are secreted by cells and constitute a significant fraction of the extracellular matrix of connective tissue. The turnover of these molecules depends on their subsequent internalization by endocytosis, their delivery to the lysosomes, and their digestion by lysosomal enzymes. The enzyme deficiencies lead to the accumulation of mucopolysaccharides in the lysosomes of the cells in the connective tissue and to an increase in their excretion in the urine. The types of mucopolysaccharidoses linked to specific enzyme deficiencies are listed below; some have been assigned an Enzyme Commission (EC) number.
Table. Types of Mucopolysaccharidoses and Associated Enzyme Deficiencies (Open Table in a new window)
Mucopolysaccharidosis Type |
Syndrome Name |
Deficiency |
EC Number |
MPS type I-H |
Hurler syndrome |
Alpha-L-iduronidase |
3.2.1.76 |
MPS type I-S (formerly MPS type V) |
Scheie syndrome |
Alpha-L-iduronidase |
N/A |
MPS type I-H/S |
Hurler-Scheie syndrome |
Alpha-L-iduronidase |
N/A |
MPS type II, mild |
Hunter syndrome, mild form |
L-sulfoiduronate sulfatase |
N/A |
MPS type II, severe |
Hunter syndrome, severe form |
L-sulfoiduronate sulfatase |
3.1.6.13 |
MPS type III-A |
Sanfilippo syndrome type A |
Heparan sulfate sulfamidase |
3.1.6.14 |
MPS type III-B |
Sanfilippo syndrome type B |
N -acetyl-alpha-D-glucosaminidase |
3.2.1.50 |
MPS type III-C |
Sanfilippo syndrome type C |
Acetyl-coenzyme A (CoA): alpha-glucosamide N -acetyltransferase |
2.3.1.3 |
MPS type III-D |
Sanfilippo syndrome type D |
N -acetyl-alpha-D-glucosamine-6-sulfatase |
3.1.6.14 |
MPS type IV-A |
Morquio syndrome, classic form |
N -acetylgalactosamine-6-sulfatase (gal-6-sulfatase) |
3.1.6.4 |
MPS type IV-B |
Morquiolike syndrome |
Beta-galactosidase |
3.2.1.23 |
MPS type VI |
Maroteaux-Lamy syndrome, mild form |
N -acetylgalactosamine-4-sulfatase (arylsulfatase B) |
N/A |
MPS type VI |
Maroteaux-Lamy syndrome, severe form |
N -acetylgalactosamine-4-sulfatase (arylsulfatase B) |
3.1.6.1 |
MPS type VII |
Sly syndrome |
Beta-glucuronidase |
3.2.1.31 |
The enzyme synthesis is controlled at the following gene loci:
-
4p16.3 (Hurler syndrome, Scheie syndrome): The activity of alpha-L-iduronidase is decreased in Hurler syndrome and Scheie syndrome. However, Hurler syndrome is a severe form of the same heavy mucopolysaccharidosis, with affected children dying after several years, whereas Scheie disease has a mild clinical phenotype. In some populations, premature stop mutations represent roughly two thirds of the mutations that cause Hurler syndrome.
-
12q14 (Sanfilippo syndrome): The diagnosis requires a specific lysosomal enzyme assay for glucosamine (N -acetyl)-6-sulfatase (GNS) activity. A homozygous nonsense mutation is found in exon 9 (1063C --> T), which predicts premature termination of translation (R355X). In addition, 2 common synonymous coding single-nucleotide polymorphisms are found and genotyped in samples from 4 ethnic groups.
-
16q24.3 (Morquio syndrome): The deficiency of enzymes in Morquio syndrome type A or type B leads to the accumulation of keratan sulfate and chondroitin-6-sulfate in the connective tissue, the skeletal system, and the teeth.
-
5q11-q13 (Maroteaux-Lamy syndrome)
-
Xq27.3-q28 (Hunter syndrome)
A new mutation has been reported, making a total of 15 different mutations that can cause premature stop codons in the alpha-L-iduronidase gene (IDUA), and the biochemistry of these mutations has been investigated. Natural stop codon read-through is dependent on the fidelity of the codon when evaluated at Q70X and W402X in CHO-K1 cells, but the 3 possible stop codons, TAA, TAG, and TGA, have different effects on mRNA stability, and this effect is context dependent.
In CHO-K1 cells expressing the Q70X and W402X mutations, the level of gentamicin-enhanced stop codon read-through is slightly less than the increment in activity caused by a lower-fidelity stop codon. In this system, gentamicin has more effect on read-through for the TAA and TGA stop codons compared with the TAG stop codon. In a mucopolysaccharidosis type I patient study, premature TGA stop codons were associated with a slightly attenuated clinical phenotype when compared with classic Hurler syndrome (eg, W402X/W402X and Q70X/Q70X genotypes with TAG stop codons). Natural read-through of premature stop codons is a potential explanation for the variable clinical phenotype in patients with mucopolysaccharidosis type I. Enhanced stop codon read-through is a potential treatment strategy for a large subgroup of patients with mucopolysaccharidosis type I.
In 25 Korean patients with Hunter syndrome, 20 mutations were identified, of which 13 mutations are novel: 6 small deletions (ie, 69_88delCCTCGGATCCGAAACGCAGG, 121-123delCTC, 500delA, 877_878delCA, 787delG, 1042_1049delTACAGCAA), 2 insertions (ie, 21_22insG, 683_684insC), 2 terminations (ie, 529G>T, 637A>T), and 3 missense mutations (ie, 353C>A, 779T>C, 899G>T). Moreover, using TaqI or HindIII restriction fragment length polymorphisms, 3 gene deletions were found. When the 20 mutations were depicted in a 3-dimensional model of iduronate 2 sulfatase protein, most of the mutations were found to be at structurally critical points that could interfere with refolding of the protein, although they were located in peripheral areas.
The candidate gene for mucopolysaccharidosis type IIIC has been localized to the pericentric region of chromosome 8 by linkage disequilibrium analysis.
Hamano et al [2] immunohistochemically examined the involvement of tauopathy/synucleinopathy, cell death, and oxidative damage in the brains of 3 cases each of mucopolysaccharidosis IIIB and mucopolysaccharidosis II and age-matched controls. In cases of mucopolysaccharidosis IIIB, the density of GABAergic interneurons in the cerebral cortex immunoreactive for calbindin-D28K and parvalbumin was markedly reduced compared with age-matched controls. The swollen neurons showed immunoreactivity for phosphorylated alpha-synuclein but not for phosphorylated tau protein or beta-amyloid protein; those in the cerebral cortex demonstrated nuclear immunoreactivity for TUNEL, single-stranded DNA and 8-OHdG. Neither lipid peroxidation nor protein glycation was marked in mucopolysaccharidosis cases. The expression levels of superoxide dismutases (Cu/ZnSOD and MnSOD) and glial glutamate transporters (EAAT1 and EAAT2) were reduced in 2 mucopolysaccharidosis II cases.
The disturbance of GABAergic interneurons can be related to mental disturbance, while synucleinopathy and/or DNA impairment may be implicated in the neurodegeneration of swelling neurons, owing to storage materials in mucopolysaccharidosis IIIB cases. These findings suggest the possibility of neuroprotective therapies other than enzyme replacement in mucopolysaccharidosis patients. [2]
The transmembrane protein gene TMEM76, which encodes a 73-kd protein with predicted multiple transmembrane domains and glycosylation sites, was found. Northern blot analysis identified 2 major TMEM76 transcripts of 4.5 kb and 2.1 kb ubiquitously expressed in various human tissues. The highest expression was detected in leukocytes and in heart, lung, placenta, and liver cells, whereas the gene was expressed at a much lower level in the thymus, colon, and brain, which is consistent with the expression patterns of lysosomal proteins. A total of 27 TMEM76 mutations were identified in the DNA of 30 mucopolysaccharidosis IIIC–affected families, which were not found in DNA from 105 controls. [3]
Functional expression of human TMEM76 and the mouse orthologue demonstrates that this gene encodes the lysosomal GNAT. Furthermore, it suggests that this enzyme belongs to a new structural class of proteins that transport the activated acetyl residues across the cell membrane. [3]
Oxidative stress may be involved in the pathophysiology of mucopolysaccharidosis type IV-A. A study of Donida et al describes an increase in oxidative damage to biomolecules (lipids, urine isoprostanes; proteins, urine di-Tyr and plasma sulfhydryl groups) and significant increases in basal DNA damage in mucopolysaccharidosis type IV-A patients. [4]
Epidemiology
Frequency
The prevalences are as follows: mucopolysaccharidosis type I-H, 1-2 cases per 100,000 population; mucopolysaccharidosis type I-S, 1 case per 250,000 population; mucopolysaccharidosis type II, 1 case per 100,000 population; mucopolysaccharidosis type III, 1 case per 25,000-75,000 population; and mucopolysaccharidosis type IV, 1 case per 40,000-200,000 population.
The prevalences of mucopolysaccharidosis types VI, VII, and I-H/S are unknown, but the prevalence of mucopolysaccharidosis type I-H/S approximates that of mucopolysaccharidosis type I-S.
According to the US National Institutes of Health, studies in Canada estimate 1 in 100,000 babies born has Hurler syndrome. The estimate for Hurler-Scheie syndrome is 1 in 115,000, and for Scheie syndrome, it is 1 in 500,000.
An epidemiologic study of the mucopolysaccharidoses in Western Australia using multiple ascertainment sources was performed and the incidence rate for the period 1969-1996 was estimated. An incidence of approximately 1 case in 107,000 live births was obtained for mucopolysaccharidosis type I-H (Hurler phenotype); 1 case in 320,000 live births (1 in 165,000 male live births) for mucopolysaccharidosis type II (Hunter syndrome); 1 case in 58,000 for mucopolysaccharidosis III (Sanfilippo syndrome); 1 case in 640,000 for mucopolysaccharidosis type IV-A (Morquio syndrome type A); and 1 case in 320,000 for mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome). The overall incidence for all types of mucopolysaccharidosis was approximately 1 case in 29,000 live births.
Murphy et al estimated the incidence (2001-2006) and prevalence (2002 census) of mucopolysaccharidosis type I in the Irish Republic (AOl) using population data. The birth incidence was 1 case in 26,206 births, with a carrier frequency of 1 case in 81 births. Of note, 19 (73%) of 26 Hurler syndrome patients were Irish Travelers. Amongst Irish Travelers, the incidence was 1 case in 371 persons, with a carrier frequency of 1 case in 10 persons. This is the highest recorded incidence worldwide. [5]
According to the incidence study covered the period from 1975-2004 in Sweden and Denmark and from 1979-2004 in Norway, the incidence of all mucopolysaccharidosis disorders was 1.75 cases in Sweden, 3.08 cases in Norway, and 1.77 cases in Denmark per 100 000 newborns. The incidence of mucopolysaccharidosis type I was the most common in all 3 countries, with 0.67, 1.85, and 0.54 cases per 100 000 newborns, respectively; for mucopolysaccharidosis type II, numbers were 0.27, 0.13, and 0.27 cases, respectively. For patients with other mucopolysaccharidosis disorders, the incidence varied widely. The prevalence for all mucopolysaccharidosis disorders was 4.24, 7.06, and 6.03 cases per million inhabitants in Sweden, Norway, and Denmark, respectively. [6]
Héron et al in the retrospective epidemiological study in France, the United Kingdom, and Greece calculated the incidence according to the number of patients born each year and then diagnosed with mucopolysaccharidosis type III before 2006. A comparison between countries focused on years 1990-1994. The calculated incidence of mucopolysaccharidosis type III in France (0.68 case per 100,000 live-births) was almost half that in the United Kingdom (1.15 cases per 100,000). Prevalence in Greece (0.97 case per 100,000 live-births) was in between France and the United Kingdom. However, mucopolysaccharidosis type IIIA was not diagnosed in Greece, and mucopolysaccharidosis type IIIB was the most highly prevalent type. [7]
Age
Onset usually occurs in early childhood.
Prognosis
Patients with Hurler syndrome usually die by age 5-10 years. The life expectancy of patients with Scheie syndrome may be nearly normal. They can live until the fifth or sixth decade of life, and they can have healthy offspring. As for patients with Hunter and Sanfilippo syndromes, death usually occurs by the time of puberty. In the classic form of Morquio syndrome, long-term survival is rare, with death occurring in persons aged 20-40 years. In patients with the severe form of Maroteaux-Lamy syndrome, death usually occurs by early adulthood.
Mucopolysaccharidosis type I (Hurler syndrome)
Patients with Hurler syndrome have a poor prognosis. Children with this disease have significant progressive physical and mental deficiencies. Death can occur in late childhood, early adolescence, or adulthood.
Mucopolysaccharidosis type II (Hunter syndrome)
The life expectancy for the early-onset form (severe form) is 10-20 years; for the late-onset form (mild form), it is 20-60 years.
Mucopolysaccharidosis type III (Sanfilippo syndrome)
Severe intellectual disability is the most important of the clinical problems. Patients may have IQs below 50. Severe cases lead to death before the patient is aged 20 years. In a minority of cases, it is compatible with a normal lifespan.
Mucopolysaccharidosis type IV (Morquio syndrome)
Bony abnormalities represent a significant problem. Small vertebrae at the top of the neck can cause slippage that damages the spinal cord, possibly resulting in paralysis. Death may occur as a result of cardiac complications.
Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome)
The life expectancy is the second to third decade of life, with patients dying from heart failure. Patients may die earlier from cardiac or neurologic complications, depending on the severity of disease.
Mucopolysaccharidosis type VII (Sly syndrome)
In mild cases, patients were reported to survive to age 19-20 years. Life expectancy is reduced as a result of frequent upper respiratory tract infections, neurodegenerative complications, and gastrointestinal tract abnormalities.
-
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
-
A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
Tables
Mucopolysaccharidosis Type |
Syndrome Name |
Deficiency |
EC Number |
MPS type I-H |
Hurler syndrome |
Alpha-L-iduronidase |
3.2.1.76 |
MPS type I-S (formerly MPS type V) |
Scheie syndrome |
Alpha-L-iduronidase |
N/A |
MPS type I-H/S |
Hurler-Scheie syndrome |
Alpha-L-iduronidase |
N/A |
MPS type II, mild |
Hunter syndrome, mild form |
L-sulfoiduronate sulfatase |
N/A |
MPS type II, severe |
Hunter syndrome, severe form |
L-sulfoiduronate sulfatase |
3.1.6.13 |
MPS type III-A |
Sanfilippo syndrome type A |
Heparan sulfate sulfamidase |
3.1.6.14 |
MPS type III-B |
Sanfilippo syndrome type B |
N -acetyl-alpha-D-glucosaminidase |
3.2.1.50 |
MPS type III-C |
Sanfilippo syndrome type C |
Acetyl-coenzyme A (CoA): alpha-glucosamide N -acetyltransferase |
2.3.1.3 |
MPS type III-D |
Sanfilippo syndrome type D |
N -acetyl-alpha-D-glucosamine-6-sulfatase |
3.1.6.14 |
MPS type IV-A |
Morquio syndrome, classic form |
N -acetylgalactosamine-6-sulfatase (gal-6-sulfatase) |
3.1.6.4 |
MPS type IV-B |
Morquiolike syndrome |
Beta-galactosidase |
3.2.1.23 |
MPS type VI |
Maroteaux-Lamy syndrome, mild form |
N -acetylgalactosamine-4-sulfatase (arylsulfatase B) |
N/A |
MPS type VI |
Maroteaux-Lamy syndrome, severe form |
N -acetylgalactosamine-4-sulfatase (arylsulfatase B) |
3.1.6.1 |
MPS type VII |
Sly syndrome |
Beta-glucuronidase |
3.2.1.31 |






