Practice Essentials
Ehlers-Danlos syndromes (EDS), the name given to a group of more than 10 different inherited, clinically and genetically heterogeneous group of connective-tissue disorders, involves a genetic defect in collagen and connective-tissue synthesis and structure. In 2017, a new international classification was proposed with 13 different variants. [1, 2]
Signs and symptoms
Also see Presentation.
To date, 11 variants of Ehlers-Danlos syndrome (EDS) are identified; all have genetic, biochemical, and clinical differences. More than one third of persons with Ehlers-Danlos syndrome do not fit exactly into a single type; overlap is common.
Common to almost all groups is a unique appearance of the skin (see the images below).
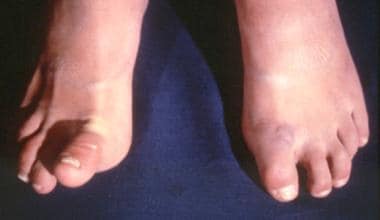 Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
 Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
 Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
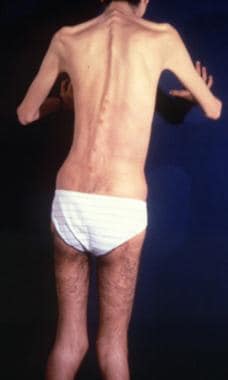 Dorsal view of a patient with Ehlers-Danlos syndrome. Note the S-curved spinal column. Courtesy of Enrico Ceccolini, MD.
Dorsal view of a patient with Ehlers-Danlos syndrome. Note the S-curved spinal column. Courtesy of Enrico Ceccolini, MD.
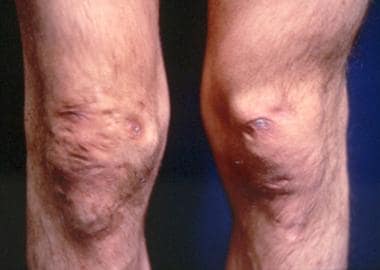 Cigarette-paper–like scars over the knees of a patient with Ehlers-Danlos syndrome. Note also the deformity of the left knee. Courtesy of Enrico Ceccolini, MD.
Cigarette-paper–like scars over the knees of a patient with Ehlers-Danlos syndrome. Note also the deformity of the left knee. Courtesy of Enrico Ceccolini, MD.
The skin is usually white and soft, and underlying vessels are sometimes visible. The skin has a doughy feel. The skin is easily hyperextensible. It is easy to pull, and, once released, it immediately returns to its original state.
Molluscoid pseudotumors are small, spongy tumors found over scars and pressure points. Molluscoid pseudotumors consist of fat surrounded by a fibrous capsule. They are commonly seen in patients with type I.
Smaller, deep, palpable, and movable nodules are often present in the subcutaneous tissue. These nodules can be found in the arms and over the tibias. Calcification leads to opacity on radiographs.
Fragility of dermal skin is common, with frequent bruises and lacerations. Poor wound healing is not rare. The use of sutures is usually a problem in patients, in whom easy dehiscence and cigarette-paper–like scars may be observed. Frequently, these scars are found on the knees.
The joints are hyperextensible, sometimes dramatically, but the degree of involvement is variable. The digit joints are most commonly affected, but all the joints can show alterations. Dislocations can occur, but patients are usually able to quickly reduce them with no pain.
Diagnostics
Calcification of small, deep, palpable, and movable nodules (often present in the subcutaneous tissue) can lead to opacity on radiographs. MRI study of the hypermobility type may document specific white matter lesions. [3]
For type IV, a prenatal diagnosis by means of polymorphic restriction genetic studies is possible.
For type VI, measurements of lysyl hydroxylase (LH) in the amniotic fluid can be used to predict the outcome of pregnancy.
In 1 patient, the vascular type of Ehlers-Danlos syndrome (EDS) was associated with mild hemophilia A. [4]
One patient with Ehlers-Danlos syndrome type IV has been diagnosed with gastric adenocarcinoma.
Histologic findings
Histologic findings in skin biopsy specimens are variable and sometimes normal. Dermal collagen fibers are disorderly arranged, with a whorled appearance. Elastic fibers show irregularities in size and orientation.
Electron microscopy reveals defects in the striations of the collagen fibers, with large or small fibrils.
Management
Also see Surgical Care and Activity.
The most common comorbidities among 1319 Danish patients with EDS, compared to a control cohort of 46,700 patients, were gastrointestinal functional disorders, hernias, asthma, pneumonia, and osteoporosis. [5] The association between some of these comorbidities and EDS may require further analysis.
Treatment is unsatisfactory.
One isolated report showed that patients with type VI disease benefited from oral vitamin C at 4 g/d. Scars and bleeding time seemed to improve with this treatment.
Rombaut et al performed a cross-sectional study of self-reported medication use, surgery, and physiotherapy in 79 patients with hypermobility-type Ehlers-Danlos syndrome (EDS) (type III), finding that 73 patients (92.4%) were taking medication, 56 (70.9%) had undergone surgery, and 41 (51.9%) were currently receiving physical therapy. [6] The patients taking medication all took analgesics; many also reported using antidepressants. Severe pain was frequent; patients taking strong opiates made more complaints and experienced greater functional impairment than those using weaker analgesics.
Rehabilitation programs specifically designed for musculoskeletal disorders of Ehlers-Danlos syndrome patients, such as a scapular motor control program with multidirectional severe shoulder instability, are desirable. [7]
A 2020 pilot study noted that custom-made foot orthoses improved foot pain, disability related to foot pain, foot function, fatigue, and mental health–related quality of life in Ehlers-Danlos syndrome patients. [8]
Background
Ehlers-Danlos syndrome can affect the skin, joints, and blood vessels. This syndrome is clinically heterogeneous and has been classically divided into 6 types (classical, hypermobile, vascular, kyphoscoliotic, arthrochalasis, and dermatosparaxis), with the underlying collagen abnormality being different for each type. Asymptomatic, nonsyndromic joint hypermobility, hypermobility spectrum disorders, and Ehlers-Danlos syndrome (particularly, the hypermobile type) are the most common phenotypes evident in association with joint hypermobility. [1] One should not regard these syndromes as curiosities and should be aware that systemic features, including chronic pain, gastrointestinal dysmotility, dysautonomia, mast cell activation and anxiety and phobic states, may be complications. [9] Advances have shown more complexities with novel, clinically overlapping, rare Ehlers-Danlos syndrome variants identified, some associated with specific genetic defects. [10] Clinical recognition of the various types of Ehlers-Danlos syndrome is important, which is particularly true for the vascular type, as it is associated with arterial rupture and visceral perforation, with possible life-threatening consequences. [11]
Pathophysiology
Ehlers-Danlos syndrome (EDS) is a heterogeneous group of inherited connective-tissue disorders characterized by joint hypermobility, cutaneous fragility, and hyperextensibility. The collagen defect has been identified in at least six of the many types of Ehlers-Danlos syndrome. The vascular form, sometimes referred to as type IV, is characterized by a decreased amount of type III collagen. It is autosomal dominant and caused by mutations in the COL3A1 gene that result in increased fragility of connective tissue with arterial, intestinal, and uterine ruptures and premature death. [12, 13] Types V and VI are characterized by deficiencies in hydroxylase and lysyl oxidase, an important posttranslational modifying enzyme in collagen biosynthesis. Type VII has an amino-terminal procollagen peptidase deficiency. Type IX has abnormal copper metabolism. Type X has nonfunctioning plasma fibronectin.
In Ehlers-Danlos syndrome types I and II, the classic variety, identifying the molecular structure in most individuals who are affected is difficult. Causative mutations may involve the COL5A1, COL5A2, and tenascin-X genes and are implied to be in the COL1A2 gene. Nonetheless, in most families with autosomal dominant Ehlers-Danlos syndrome, the disease appears to be linked to loci that contain the COL5A1 or COL5A2 genes. Although half of the mutations that cause Ehlers-Danlos syndrome types I and II are likely to affect the COL5A1 gene, a significant portion of the mutations result in low levels of mRNA from the mutant allele as a consequence of nonsense-mediated mRNA decay. [14] Among 31 female and 13 male Polish patients with classic Ehlers-Danlos syndrome, nine new mutations of the COL5A1 gene were delineated (8 missense mutations and 1 splice site). [2]
Bouma et al evaluated 3 generations in a family with Ehlers-Danlos syndrome type II. [15] The genomic defect was an A(-2)→G substitution at the exon 14 splice-acceptor site. Transmission electron micrographs of type I collagen fibrils in a proband dermal biopsy specimen demonstrated heterogeneity in fibril diameter that was greater than that of a matched control sample. The proband was found to have a greater proportion of both larger and smaller fibrils, and occasional fibrils with a cauliflower configuration were observed. [15]
Wenstrup and associates identified haploinsufficiency of the COL5A1 gene that encodes the proalpha1(V) chain of type V collagen in the classic form of Ehlers-Danlos syndrome. Eight of 28 probands with classic Ehlers-Danlos syndrome who were heterozygous for expressed polymorphisms in COL5A1 had complete or nearly complete loss of expression of one COL5A1 allele. One third of individuals with classic Ehlers-Danlos syndrome were estimated to have mutations of COL5A1 that result in haploinsufficiency. These findings suggest that the normal formation of the heterotypic collagen fibrils that contain types I, III, and V collagen requires the expression of both COL5A1 alleles. [16] Type V collagen mutations are pivotal in classic Ehlers-Danlos syndrome. [17]
Autosomal recessive–type VI Ehlers-Danlos syndrome, also known as the kyphoscoliotic type, is characterized by neonatal kyphoscoliosis, generalized joint laxity, skin fragility, and severe muscle hypotonia at birth. Biochemically, this type is attributed to a deficiency in lysyl hydroxylase (LH), the enzyme that hydroxylates specific lysine residues in the collagen molecule to form hydroxylysines with two important functions. The residues are attachment sites for galactose and glucosylgalactose, and they act as precursors of the cross-linking process that gives collagen its tensile strength.
More than 20 mutations are identified in the LH1 gene that contributes to LH deficiency and clinical Ehlers-Danlos syndrome type VI. Yeowell and Walker identified two of these mutations in five or more unrelated patients: (1) a large duplication of exons 10-16, which arise from a homologous recombination of intronic Alu sequences, and (2) a nonsense mutation, Y511X, in exon 14 of the LH1 gene. Both mutations seem to originate from a single ancestral gene. [18]
Tenascin-X is a large extracellular matrix protein, a deficiency of which causes a clinically distinct recessive form of this syndrome. [19] Thus, factors other than collagens or collagen-processing enzymes may cause this syndrome. This newly described form may be associated with additional anomalies.
A case with colonic perforation in a young girl, with a fatal outcome, was related to a novel mutation of the COL3A1 gene. Crystal structure of human type III, in the structure G991-G1032 cystine knot, shows both 7/2 and 10/3 symmetries. [20]
Vascular Ehlers Danlos syndrome (vEDS), characterized by arterial aneurysm, dissection and rupture, bowel rupture, and rupture of the gravid uterus, results from pathogenic variants in COL3A1, which encodes the chains of type III procollagen. [11] Pathogenic variants in COL3A1, most frequently glycine substitutions, with glutamic acid to lysine substitutions (Glu>Lys) in COL3A1, also were delineated. [13] A novel point mutation has been found in the vascular type of Ehlers-Danlos syndrome. The mutation occurs in the second position of exon 24 of COL3A1. [11, 21]
Impaired wound healing is a typical feature of Ehlers-Danlos syndrome, probably for a fibroblast defect. Wound repair can be achieved using exogenous type V collagen. [22]
Ehlers-Danlos syndrome pediatric patients have been shown to have deficiencies in three genes of the glutathione S-transferase family (GSTM1, GSTT1, GSTP1). This leads to the generation of reactive oxygen species. [23]
Reduced activity of beta4-galactosyltransferase 7 (beta4GalT-7) is associated with the progeriform Ehlers-Danlos syndrome.
Biallelic mutations in FKBP14 may result in a recessive form of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, hearing loss, and, possibly, an increased risk for vascular complications. [24]
Etiology
Collagens are a family of proteins that are widely distributed in all organs of the body. Thirteen different subtypes are known, and the number increases constantly. The joints, blood vessels, and skin have different kinds of collagen in their structure; in all these locations, collagen is organized into bundles.
Collagen organization is not easily visible by means of light microscopy, and abnormalities are better detected with electron microscopy. Collagen disorganization and/or abnormal bundle size is correlated with clinical evidence in the joints, skin, and blood vessels.
In Ehlers-Danlos syndrome (EDS), skin collagen alteration can be seen in the reticular dermis. Two abnormalities are evident: irregularities in the diameter of the fibrils and irregular collagen shapes. Fibrils can be large and irregular in some types (types I-III), but they can also be small or varied in others. The most severe form of Ehlers-Danlos syndrome, type IV, is the best studied; biochemically, this type has decreased or absent type III collagen synthesis. Pathologic findings in other skin layers are visible but nonspecific.
Epidemiology
Frequency
The prevalence of Ehlers-Danlos syndrome (EDS) is reported to be 1 case in approximately 400,000 people, but mild or incomplete forms appear to be underdiagnosed and more common than other forms. The frequency of vascular Ehlers-Danlos syndrome has been estimated at 1 case in 50,000 to 1 case in 200,000. [11]
Race-, sex-, and age-related information
No racial predominance seems to exist; however, some believe that Whites probably are affected more than other races.
The sex-related prevalences are almost equal.
The disease has clinical features (eg, joint mobility, skin extendibility, scarring tendency) that are easily recognizable beginning in early childhood. The other clinical manifestations require more time to become evident. Ehlers-Danlos syndrome is usually diagnosed in young adults.
Prognosis
Type IV Ehlers-Danlos syndrome (EDS) is a severe form. Patients often have a shortened lifespan because of the spontaneous rupture of a large artery (eg, splenic artery, aorta) or the perforation of internal organs. Surgery can pose life-threatening risks in these patients. [25] Arterial aneurysms, valvular prolapse, and spontaneous pneumothorax are common complications. The prognosis with this type is poor. Sudden death can occur after visceral perforation or after the rupture of a large vessel, most commonly an abdominal and splenic vessel.
The other types are usually not as dangerous, and affected individuals can live a healthy if somewhat restricted life. Type VI is also somewhat dangerous, although it is rare.
-
Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
-
Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
-
Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
-
Dorsal view of a patient with Ehlers-Danlos syndrome. Note the S-curved spinal column. Courtesy of Enrico Ceccolini, MD.
-
Cigarette-paper–like scars over the knees of a patient with Ehlers-Danlos syndrome. Note also the deformity of the left knee. Courtesy of Enrico Ceccolini, MD.
-
Criteria for Ehlers-Danlos syndrome are shown in Media Files 6-11. Dorsiflexion of the little finger by more than 90°with the forearm flat on the table.
-
Passive apposition of the thumb to the flexor forearm.
-
Hyperextension of the elbow by more than 90°.
-
Hyperextension of the knee by more than 10°.
-
Forward flexion of the trunk until the palms of the hands rest easily on the floor.
-
Evaluation of skin extensibility.







