Practice Essentials
Holoprosencephaly denotes an incomplete or absent division of the embryonic forebrain (prosencephalon) into distinct lateral cerebral hemispheres. [1, 2, 3, 4, 5, 6] DeMyer historically and roughly categorized holoprosencephaly into 3 types (from most severe to least severe) [7] :
-
Alobar holoprosencephaly
-
Semilobar holoprosencephaly
-
Lobar holoprosencephaly
In alobar holoprosencephaly (shown in the image below), there is a complete absence of midline forebrain division, resulting in a monoventricle and fused cerebral hemispheres.
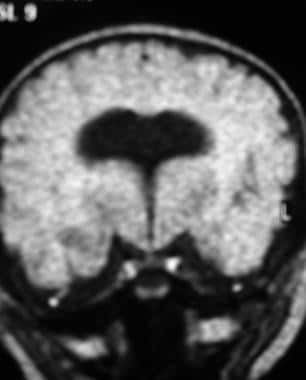 Coronal fluid-attenuated inversion recovery magnetic resonance image shows alobar holoprosencephaly.
Coronal fluid-attenuated inversion recovery magnetic resonance image shows alobar holoprosencephaly.
Semilobar holoprosencephaly is characterized by an incomplete forebrain division, resulting in partial separation of the cerebral hemispheres, typically posteriorly.
In lobar holoprosencephaly (seen in the image below), there is complete ventricular separation, with focal areas of incomplete cortical division or anterior falcine hypoplasia present.
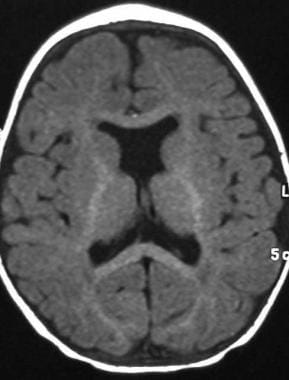 Axial T1-weighted magnetic resonance image shows septo-optic dysplasia, representing a variant of lobar holoprosencephaly.
Axial T1-weighted magnetic resonance image shows septo-optic dysplasia, representing a variant of lobar holoprosencephaly.
Distinctive midline facial malformations occur in most cases. Typical facial anomalies are correlated with the degree of holoprosencephaly and have prognostic importance. [2, 3] From most severe to least severe, these include the following: (1) cyclopia, in which a single, midline, fused eye exists in a single orbit below a proboscis; (2) ethmocephaly, in which ocular hypotelorism is present with an interorbital proboscis; (3) cebocephaly, in which ocular hypotelorism is present with a single-nostril nose; (4) ocular hypotelorism and midline clefting; and (5) ocular hypotelorism and bilateral clefting.
More subtle facial dysmorphic features may also be present. These include a flat nasal bridge and tip; a single, midline, upper incisor; a bifid uvula; absent nasal bones and nasal septum; and congenital nasal pyriform aperture stenosis (decreased width of the nasal pyriform aperture at the level of the interior meatus).
Microcephaly is the rule, and macrocephaly, if present, is suggestive of hydrocephalus.
Virtually all surviving individuals with the more severe forms of holoprosencephaly have some developmental delay, often persisting as intellectual disability. In general, this finding is directly correlated with the severity of holoprosencephaly.
Also not uncommon are seizures, hypotonia and/or hypertonia, extrapyramidal features, such as dystonia and/or chorea; hypothalamic and brainstem dysfunction leading to autonomic dysfunction and swallowing difficulties; pituitary dysfunction, which can manifest as partial or complete panhypopituitarism with resultant endocrine deficiencies; and feeding difficulties, which can lead to aspiration pneumonia and failure to thrive.
Active research into the pathophysiology of holoprosencephaly has revealed multiple teratogenic and genetic causes (chromosomal and single gene); further genetic characterization is ongoing.
Preferred examination
The imaging study of choice for the diagnosis and classification of holoprosencephaly is cranial magnetic resonance imaging (MRI). [8, 1, 9, 10, 11] The next best imaging modalities are ultrasonography and cranial computed tomography (CT) scanning. [2, 3, 12, 13, 14, 15, 16] Ultrasonography can be limited in cases of microcephaly if there is a very small fontanelle. Transcranial ultrasonography, however, can be performed through the calvaria, at times using the thinner temporal bones. Often this technique requires a transducer of lower frequency, resulting in better penetration, albeit at the loss of some near-field resolution.
Prenatal evaluation by means of transabdominal or transvaginal ultrasonography can be used to identify most cases of alobar or semilobar holoprosencephaly. Prenatal MRI can be done in cases in which the infant's head is not easily accessible at the time of ultrasonographic evaluation or in cases in which the anatomy is not satisfactorily delineated at prenatal ultrasonography. [1]
Prenatal ultrasonography is not a reliable method for diagnosing mild forms of holoprosencephaly, such as lobar holoprosencephaly, because of its high false-negative rate. In addition, prenatal ultrasonography often cannot distinguish between alobar holoprosencephaly and semilobar holoprosencephaly. A transabdominal ultrasonographic diagnosis of holoprosencephaly before 16 weeks' gestation is difficult. [17, 18, 1]
A study of 35 fetuses with holoprosencephaly compared the accuracy of the diagnosis between ultrasound and in utero MRI. There were 9 false negative in utero MRI findings. Of the 26 cases of holoprosencephaly diagnosed on in utero MR imaging, 12 were not recognized on ultrasonography. [19]
Radiography
Craniofacial defects associated with holoprosencephaly that may be detected on plain radiographs include a single, fused orbit; orbital hypotelorism; abnormal nasal bone formation; facial clefts; and a single, midline, central incisor.
Changes apparent on plain radiography are nonspecific and are insufficient to make the diagnosis of holoprosencephaly.
Not all individuals with holoprosencephaly have craniofacial abnormalities that are detectable on plain radiography. However, holoprosencephaly is in the differential diagnosis whenever these associated anomalies are found.
Computed Tomography
CT scans can establish a diagnosis of holoprosencephaly by providing images of brain anatomy. CT scanning is best suited for imaging the bony structure of the skull. However, CT scanning exposes the patient to ionizing radiation and is therefore relatively contraindicated in the prenatal diagnosis. CT scanning provides less detail of the brain parenchyma than MRI does, and it does not provide good images of the posterior fossa and brainstem. Therefore, some cases of mild holoprosencephaly and associated central nervous system (CNS) anomalies can be missed on CT scans.
On CT scan images, alobar holoprosencephaly results in a horseshoe-shaped monoventricle, an absent interhemispheric fissure, fused thalami, an absent falx, agenesis of the corpus callosum, an absent septum pellucidum, and absent olfactory bulbs.
Semilobar holoprosencephaly is characterized by partial ventricular differentiation, but with a single ventricular cavity, a partial interhemispheric fissure and falx (posterior-ventral axis), partial or incomplete formation of the corpus callosum, and a variable degree of thalamic fusion. The olfactory bulbs are often absent. The abnormality is more severe anteriorly, with partial cleavage and lateral differentiation occurring posteriorly. In patients with semilobar holoprosencephaly, the posterior portion of the corpus callosum is present, and the more anterior portion, where failure of cleavage has occurred, is absent.
Lobar holoprosencephaly occurs with partial fusion of the frontal lobe, with an otherwise normally formed interhemispheric fissure, lateral ventricular formation, variable and incomplete absence of the anterior corpus callosum and/or septum pellucidum, and separate thalami. The olfactory tracts are present.
The middle interhemispheric fusion variant appears as incomplete cleavage of the posterior frontal and parietal lobes and, often, incomplete cleavage of the basal ganglia and thalami. The body of the corpus callosum is absent in the area where cleavage has failed to occur. [20]
Magnetic Resonance Imaging
Cranial MRI is the diagnostic imaging modality of choice in cases of holoprosencephaly. [19, 8, 1] Alobar holoprosencephaly results in a horseshoe-shaped monoventricle, an absent interhemispheric fissure, fused thalami, an absent falx, agenesis of the corpus callosum, an absent septum pellucidum, absent olfactory bulbs, abnormal cerebral cortex, and migration anomalies. The MRI characteristics of alobar holoprosencephaly are demonstrated in the images below.
Coronal fluid-attenuated inversion recovery magnetic resonance image shows alobar holoprosencephaly.
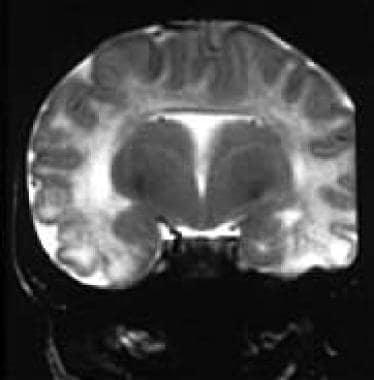 Fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
Fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
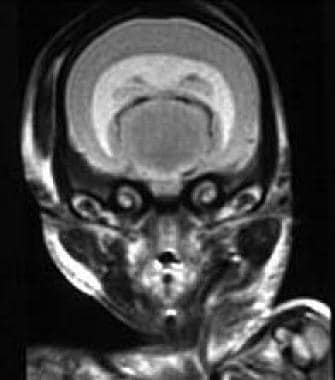 Early fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
Early fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
Semilobar holoprosencephaly is characterized by partial ventricular differentiation, but with a single ventricular cavity, a partial interhemispheric fissure and falx (posterior-ventral axis), partial or incomplete formation of the anterior corpus callosum, and a variable degree of thalamic fusion. The olfactory bulbs are often absent.
Lobar holoprosencephaly (seen in the image below) occurs with partial fusion of the frontal lobe, with an otherwise normally formed interhemispheric fissure, lateral ventricular formation, variable and incomplete absence of the anterior corpus callosum and/or septum pellucidum, and separate thalami. The olfactory tracts are present.
Axial T1-weighted magnetic resonance image shows septo-optic dysplasia, representing a variant of lobar holoprosencephaly.
The middle interhemispheric fusion variant appears as incomplete cleavage of the posterior frontal and parietal lobes and incomplete cleavage of the basal ganglia and thalami. The body of the corpus callosum is absent, coincident with the site of failure of cerebral separation. [20]
Diffusion tensor imaging (DTI) has been reported to offer a better analysis of cerebral white matter connectivity than conventional MRI and identify microstructural changes that occur in patients with the middle interhemispheric variant of holoprosencephaly. [21]
Fetal MRI may yield false-positive or false-negative results, and fetal activity can significantly degrade the images and may need to be circumvented by repeated sequences and single-shot fast spin-echo (SS-FSE) MRI techniques.
Ultrasonography
On preenatal ultrasonography, primary CNS findings include the following: (1) single, sickle-shaped or horseshoe monoventricle; (2) absent midline echo due to the absence of an interhemispheric fissure, falx, corpus callosum, and septum pellucidum; (3) thin cortical rim; (4) single, fused thalamus; (5) microcephaly; and (6) ventriculomegaly/hydrocephalus. [12, 17, 18]
Associated craniofacial ultrasonographic findings include the following: (1) ocular hypotelorism or cyclopia, (2) proboscis or abnormal nasal bone formation, and (3) cleft lip and/or palate (midline or bilateral).
Postnatally, alobar holoprosencephaly shows a single ventricle, fused thalami, and a thin, usually poorly differentiated cortical mantle. A dorsal cyst or ventricular remnant may also be detected.
Semilobar holoprosencephaly is detected by identifying the partial cleavage of the occipital horns and the presence of a posterior falx and a posterior portion of the corpus callosum. Anteriorly, there is typically fusion of the ventricles and of the hemispheres, with concomitant absence of the corpus callosum and of the septum pellucidum.
Lobar holoprosencephaly is a continuum. In some patients, the ultrasonographic images show fusion of the anterior horns and of portions of the frontal lobes. The mildest forms of lobar holoprosencephaly may be manifested only by absence of the septum pellucidum.
Sonograms of the middle interhemispheric fusion variant demonstrate normal cleavage anteriorly and posteriorly, with fusion of the hemispheres and absence of the body of the corpus callosum in the posterior, frontal, and parietal regions. [20]
Degree of confidence
Reportedly, cases of alobar holoprosencephaly have been detected as early as 9-14 weeks' gestation (and often detected at 18-20 weeks on routine anatomic scans), semilobar holoprosencephaly has been detected by 13-20 weeks' gestation, and lobar holoprosencephaly has been detected by 21 weeks' gestation with the use of transvaginal ultrasonography. Conventional transabdominal ultrasonography cannot achieve this degree of early detection.
Fetal MRI should be considered to confirm and further classify cases of holoprosencephaly.
Prenatal ultrasonography is not a reliable method of diagnosing mild forms of holoprosencephaly, such as lobar holoprosencephaly, because of its high false-negative rate. In addition, prenatal ultrasonography often cannot distinguish between alobar holoprosencephaly and semilobar holoprosencephaly. Transabdominal ultrasonographic diagnosis of holoprosencephaly before 16 weeks of gestation is difficult.
A false-positive diagnosis of holoprosencephaly has been reported in cases of hydrocephalus, hydranencephaly, arachnoid or porencephalic cysts, Dandy-Walker malformations with ventriculomegaly, septo-optic dysplasia, and other CNS malformations.
-
Coronal fluid-attenuated inversion recovery magnetic resonance image shows alobar holoprosencephaly.
-
Fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
-
Early fetal magnetic resonance image shows alobar holoprosencephaly. Courtesy of Dorothy I. Bulas, MD.
-
Axial T1-weighted magnetic resonance image shows septo-optic dysplasia, representing a variant of lobar holoprosencephaly.
-
Presumed alobar holoprosencephaly in a kitten.



